

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The cyclic AMP (3′, 5′-cyclic adenosine monophosphate, cAMP) is a second messenger formed by adenylate cyclase following the stimulation of cells by many neurotransmitters and hormones. cAMP activates effector molecules such as cAMP-dependent protein kinase (protein kinase A, PKA), exchange protein directly activated by cAMP, and cyclic nucleotide-gated ion channels. The effector molecules control a variety of physiological phenomena, including energy metabolism, gene expression, differentiation, proliferation, and apoptosis.1 Abnormal alteration in cAMP signaling can result in various diseases, including endocrine disorders and cancer,2 and cAMP signaling is a potential target for developing new therapy for related diseases.3
Genomic DNA continuously encounters DNA injuries caused by various exogenous or endogenous insults, such as ionizing radiation, chemicals, and stalled replication. When DNA damage is not repaired correctly by cellular repair systems, they can cause mutations, chromosomal aberrations, aneuploidy, and cell death.4 The misrepaired DNA may induce cell death and numerous diseases, such as cancer and neurological disease.5 The most serious DNA injuries are DNA double-strand breaks (DSBs), and DSBs are fixed mainly by non-homologous end joining (NHEJ) across the cell cycle and homologous recombination at the S/M phase of cell cycle.6
cAMP signaling was found to be involved in DNA repair and the development of drug resistance to chemotherapeutic agents,7 to enhance the repair of cyclobutane pyrimidine dimers induced by ultraviolet (UV) radiation8 and to increase the levels of enzymes for base excision repair.9 cAMP signaling was also described to inhibit the base excision repair of 8-oxo-deoxyguanosine induced by γ-ray irradiation10 and the NHEJ repair of DNA DSBs resulted from γ-radiation.11 However, the effect of cAMP on DNA repair is not consistent in the literature, and the mechanism underlying the different effects of cAMP remains unclarified. The different effects of cAMP signaling on DNA repair may result from the differences in factors such as DNA damaging agents, type of DNA damage, DNA repair mechanisms, and cell types. This study aimed to investigate the different mechanisms by which cAMP signaling modulates DNA repair by analyzing the cell-type-specific differences in the modulation of DNA repair by cAMP signaling following γ-ray irradiation.
METHODS
Cells, cell culture, and reagents
Human malignant melanoma cells (SK-MEL-28, male; SK-MEL-2, sex unknown), uterine cervical cancer cells (HeLa, female) and non-small cell lung cancer cells (A549, male; H1299, sex unknown) were obtained from the Korea Cell Line Bank (Seoul, Korea). SiHa human cervical cancer cells (female) were a gift of Dr. HD Youn (Seoul National University, Seoul, Korea). HeLa, SiHa and H1299 cells were grown in Dulbecco's modified Eagle's medium (DMEM), SK-MEL-2 and A549 cells were cultured in RPMI 1640, and SK-MEL-28 cells were cultured in Minimum Essential Medium (MEM). All the media contained 10% fetal bovine serum (Welgene, Gyeongsan, Korea) and 100 units/mL of penicillin/streptomycin (Welgene), and the cells were incubated at 37°C in a 5% CO2 incubator.
Prostaglandin E2 (PGE2) was purchased from Cayman Chemical (Ann Arbor, MI, USA), and okadaic acid was obtained from Tocris Bioscience (Bristol, UK). Isoproterenol, H-89, dimethyl sulfoxide and 4′, 6-diamidino-2-phenylindole (DAPI) were purchased from Sigma-Aldrich (St. Louis, MO, USA). N6-phenyladenosine-3′, 5′-cyclic monophosphate (6-Phe-cAMP) was purchased from the Biological Life Science Institute (Bremen, Germany), and bovine serum albumin was obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
Transfection and western blot analysis
Cells were transfected with expression plasmids using Lipofectamine 3000 (Invitrogen, CA, USA), and were exposed to γ-rays (5 Gy) as previously described.11 Antibodies against phosphorylated cAMP response element bind protein (p-CREB, Ser139), phosphorylated ataxia telangiectasia-mutated protein (p-ATM, Ser1981), and the catalytic subunit of DNA-dependent protein kinase (DNA-PKcs) were obtained from Santa Cruz Biotechnology. Antibodies against p-H2AX (Ser133), phosphorylated PKA substrates, cleaved poly (ADP-ribose) polymerase (PARP), and cleaved caspase-9 were purchased from Cell Signaling Technology (Beverly, MA, USA). An antibody against β-actin was purchased from Sigma-Aldrich, and antibodies against DNA-PKcs phosphorylated at Thr2609 (T2609) and Ser2056 (S2056) were purchased from Abcam (Cambridge, UK). The proteins were visualized using enhanced chemiluminescence (Thermo Fisher Scientific, Waltham, MA, USA), and the blot images were recorded using the LAS-3000 luminescent image analyzer (Fuji, Tokyo, Japan). The densities of the visualized bands were analyzed using Multi Gauge v.2.3 software (Olympus, Tokyo, Japan), and the band densities were expressed as ratios of the corresponding control densities.
Statistical analysis
All of the experiments were repeated at least three times independently, and the data were expressed as means and standard error. Statistical significance of the difference was analyzed using the nonparametric Mann-Whitney U test, and a P value ≤ 0.05 was considered statistically significant.
RESULTS
cAMP signaling modulates γ-ray-induced DNA damage and apoptosis differently depending upon the cell type
To explore the effect of cAMP signaling following DNA damage upon different cells, DNA damage following γ-ray irradiation after the activation of cAMP signaling was analyzed using melanoma cells, lung cancer cells, and uterine cervical cancer cells. Activation of cAMP signaling by pretreating melanoma cells (SK-MEL-2 and SK-MEL-28) and uterine cervical cancer cells (HeLa and SiHa) with PGE2 or isoproterenol decreased the levels of γ-H2AX, a biomarker for DNA damage, resulted from γ-ray irradiation (Fig. 1A and B). By contrast, activation of cAMP signaling in lung cancer cells (A549 cells and H1299 cells) augmented γ-H2AX levels (Fig. 1C). Thus, cAMP signaling has different effects on radiation-induced DNA damage depending upon the cell type.
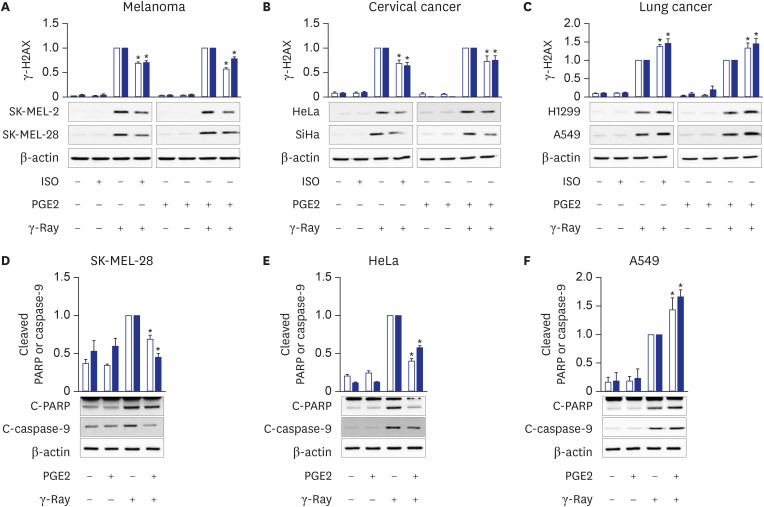
Fig. 1
Cell-type-specific modulation of γ-ray-induced DNA damage and apoptosis by cAMP signaling. (A) Effects of pretreatment with isoproterenol and PGE2 on γ-H2AX formation following γ-ray irradiation in human malignant melanoma cells. The empty bars present SK-MEL-2 cells, and the filled bars present SK-MEL-28 cells. (B) Effects of PGE2 and isoproterenol on γ-H2AX formation following γ-ray irradiation in human uterine cervical cancer cells. The empty bars present HeLa cells, and the filled bars present SiHa cells. (C) Effects of PGE2 and isoproterenol on γ-H2AX formation following γ-ray irradiation of human non-small cell lung cancer cells. The empty bars present H1299 cells, and the filled bars present A549 cells. The cells pretreated with 1 μM isoproterenol (ISO) or 20 μM PGE2 for 30 minutes were irradiated with γ-rays (5 Gy) and collected with a cell scraper after 1 h for analysis by western blotting. (D, E, F) PGE2 effects on the cleavage of PARP and caspase-9 following γ-ray irradiation in SK-MEL-28 cells (D), HeLa cells (E), and A549 cells (F). The cells pretreated with PGE2 (20 μM, 30 minutes) were irradiated with γ-rays (5 Gy) and collected with a cell scraper after 24 hours (A549 and HeLa) or 48 hours (SK-MEL-28) for analysis by western blotting. The empty bars shows cleaved PARP, and the filled bars shows cleaved caspase-9. The means ± standard error calculated from the three independent experiments were presented as columns. The asterisk (*) denotes statistically significant differences compared with the respective control (P ≤ 0.05, Mann-Whitney U test).
![]()
Next, we explored the effect of cAMP signaling on apoptosis caused by exposure to γ-rays in different cells. Activation of cAMP signaling in SK-MEL-28 melanoma cells and HeLa uterine cervical cancer cells with PGE2 reduced the cleavage of PARP and caspase-9 following γ-ray irradiation (Fig. 1D and E). Activation of cAMP signaling following the same treatment of A549 lung cancer cells augmented the cleavage of PARP and caspase-9 following irradiation (Fig. 1F). The results indicate that the cell-type-specific effects of cAMP signaling on DNA damage might result in the corresponding cell-type-specific effects on apoptosis.
cAMP signaling modulates the NHEJ repair of DNA damage resulted from γ-ray irradiation differently depending upon the cell type
To examine whether the different effects of cAMP signaling following DNA damage depending upon the cell types results from the modulation of DNA damage repair, the cAMP effect upon the disappearance of DNA damage following γ-ray irradiation was examined. Activation of cAMP signaling of SK-MEL-28 cells and HeLa cells by treating PGE2 at10 minutes after γ-ray irradiation accelerated the disappearance of γ-H2AX (Fig. 2A and B), but the same pretreatment of A549 cells delayed the disappearance of γ-H2AX (Fig. 2C). This finding indicates that the cell-type-specific effect of cAMP signaling upon DNA damage might result from the cell-type-specific modulation of cAMP signaling upon DNA repair.
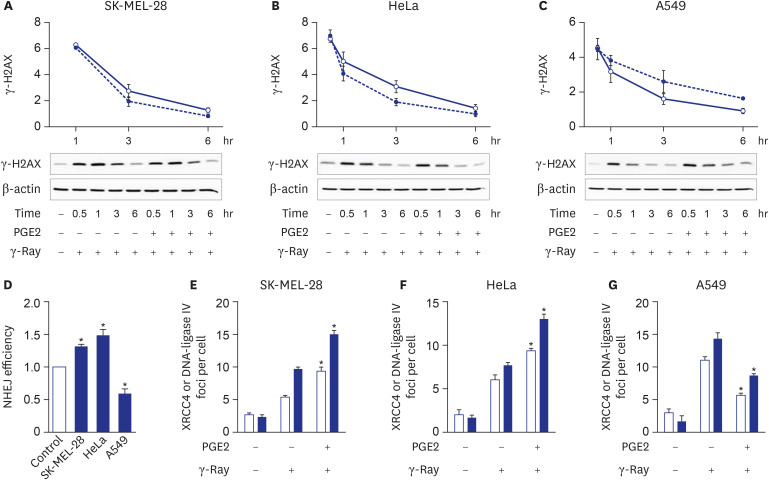
Fig. 2
Cell-type-specific modulation of NHEJ repair by cAMP signaling. (A, B, C) Effects of cAMP signaling upon the disappearance of γ-H2AX formed by γ-ray irradiation in SK-MEL-28 cells (A), HeLa cells (B), and A549 cells (C). The cells were irradiated with γ-ray, then incubated for 10 min before PGE2 treatment and collected at the presented time points for analysis by western blotting. The empty circles present vehicle-stimulated control cells, and the filled circles present PGE2- stimulated cells. (D) cAMP signaling effects upon NHEJ repair efficiency. The cells were cotransfected with GαsQL and SceI-linearized GFP fluorescent reporter plasmids using Lipofectamine 3000 and harvested after 24 hours for flow cytometric analysis. The NHEJ repair efficiencies were determined as the ratios of green fluorescence emitted from the repaired reporter plasmid to the red fluorescence emitted from the DsRed control plasmid. (E, F, G) PGE2 effects upon the recruitment of XRCC4 and DNA-ligase IV after exposure to γ-rays of SK-MEL-28 cells (E), HeLa cells (F), and A549 cells (G). The empty bars present XRCC4, and the filled bars present DNA-ligase IV. After pretreatment with PGE2, the cells were irradiated with γ-rays. The irradiated cells were reacted with antibodies against XRCC4 or DNA-ligase IV, followed by DAPI staining. The number of γ-H2AX foci per cell was derived by analyzing the confocal images of stained cells. The means ± standard error calculated from the three independent experiments were presented as columns. The asterisk (*) denotes statistically significant differences compared with the respective control (P ≤ 0.05, Mann-Whitney U test).
![]()
Next, the effect of cAMP signaling upon NHEJ activity, a major mechanism for DNA DSB repair, was tested in different cell types. Activation of cAMP signaling by expressing a constitutively active mutant of the long-form stimulatory α subunit of GTP-binding protein (GαsQL) in SK-MEL-28 and HeLa cells increased NHEJ efficiency assessed using fluorescent reporter plasmids, but the expression of GαsQL in A549 cells decreased NHEJ efficiency in the fluorescent reporter assay (Fig. 2D and Supplementary Fig. 1).
Then, to study how cAMP signaling modulates NHEJ differently, the recruitment of XRCC4 and DNA-ligase IV, key molecules in NHEJ, following γ-ray irradiation after stimulation of cAMP signaling was examined. Pretreatment with PGE2 augmented the foci formation of XRCC4 and DNA-ligase IV at DSB sites in SK-MEL-28 and HeLa cells (Fig. 2E and F; Supplementary Figs. 2 and 3), but the same pretreatment decreased the foci formation in A549 cells (Fig. 2G; Supplementary Fig. 4).
These results show that cAMP signaling exerts cell-type-specific effects on the NHEJ repair of DNA damage following exposure to γ-rays.
cAMP signaling regulates the phosphorylation of DNA-PKcs cell-type specifically following exposure to γ-rays
To probe the mechanisms by which cAMP signaling modulates NHEJ repair differently, we analyzed the effects of cAMP signaling upon the phosphorylation of DNA-PKcs, the catalytic subunit of DNA-dependent protein kinase (DNA-PK), a major enzyme in NHEJ. The DNA-PKcs phosphorylation is implied to regulate enzyme activation, processing of DNA ends, and DNA ligation. Activation of cAMP signaling in SK-MEL-28 melanoma and HeLa uterine cervical cancer cells by pretreatment with PGE2 increased the phosphorylation of DNA-PKcs on T2609 and the activating phosphorylation of ATM on S1981 (Fig. 3A and 3). Pretreatment of SK-MEL-28 and HeLa cells with okadaic acid, a protein phosphatase 2A inhibitor, further increased the PGE2-induced phosphorylation of DNA-PKcs on T2609 and ATM on S1981 (Fig. 3A and 3). By contrast, the activation of cAMP signaling in A549 lung cancer cells using PGE2 decreased both the phosphorylation of DNA-PKcs on T2609 and ATM on S1981 (Fig. 3C). The effects of cAMP on the phosphorylation of DNA-PKcs on T2609 and phosphorylation of ATM on 1981 were abolished by treatment with okadaic acid in A549 cells (Fig. 3C). The results shows that cAMP signaling decreases the phosphorylation of DNA-PKcs on T2609 through PP2A- and ATM-dependent pathways in lung cancer cells but increases the phosphorylation of DNA-PKcs on T2609 through PP2A-independent pathways in melanoma and cervical cancer cells. Additionally, the activation of cAMP signaling by treatment with PGE2 reduced the γ-ray-induced phosphorylation of DNA-PKcs on S2056 in SK-MEL-28 and HeLa cells but increased the phosphorylation in A549 cells (Fig. 3D and E).
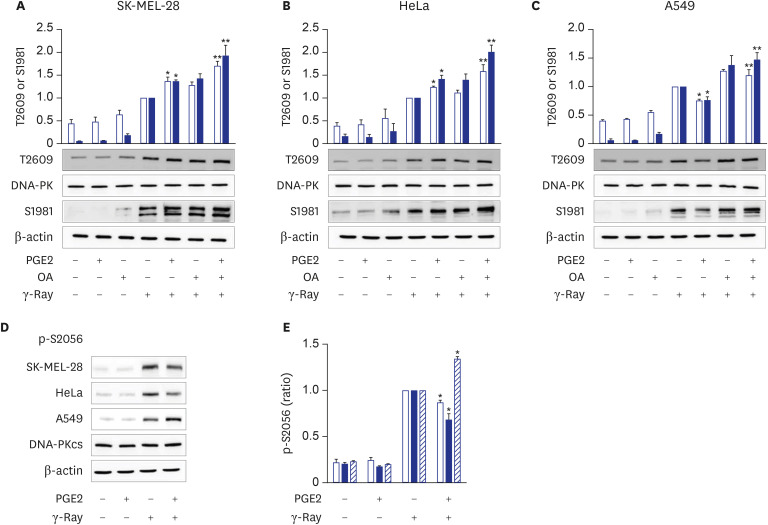
Fig. 3
Cell-type-specific modulation of DNA-PKcs phosphorylation at T2609 and S2056 following γ-ray irradiation by cAMP signaling. (A, B, C) PGE2 effects upon the phosphorylation of DNA-PKcs on T2609 and ATM on S1981 following γ-ray irradiation in SK-MEL-28 cells (A), HeLa cells (B), and A549 cells (C). The cells were treated with PGE2 with/without pretreatment with okadaic acid (100 nM, 30 minutes). The treated cells were exposed to γ-rays and collected after 1 hour for analysis by western blotting. The empty bars present the phosphorylation of DNA-PKcs at T2609, and the filled bars present the phosphorylation at ATM on S1981. (D) PGE2 effects upon the phosphorylation of DNA-PKcs at S2056 following exposure to γ-rays. (E) Bar graph obtained by densitometric analysis of images in (D). The cells were irradiated with γ-rays after pretreatment with PGE2, and the irradiated cells were collected for analysis by western blotting. The empty bars present SK-MEL-28 cells, the filled bars present HeLa cells, and the slant bars present A549 cells. The means ± standard error calculated from the three independent experiments were presented as columns. An asterisk (*) denotes statistically significant differences compared with the irradiated control, and double asterisk (**) denotes differences indicating statistical significance compared with the PGE2- stimulated cells (P ≤ 0.05, Mann-Whitney U test).
![]()
These results shows that cAMP signaling modulates the radiation-induced phosphorylation of DNA-PKcs differently depending upon the cell type, suggesting that the cell-type-specific modulation of NHEJ repair by cAMP signaling might be mediated by the cell-type-specific modulation of cAMP signaling by DNA-PKcs phosphorylation.
PKA mediates the cell-type-specific phosphorylation of DNA-PKcs by cAMP signaling following exposure to γ-rays
To probe the molecules that mediate the cell-type-specific modulation of NHEJ repair by cAMP, the role of PKA, a major effector of cAMP signaling, was analyzed. Inhibition of PKA by treating with H-89, a PKA-specific inhibitor, abolished both the stimulatory effect of PGE2 upon the phosphorylation on T2609 induced by γ-ray irradiation in HeLa cells and SK-MEL-28 cells and the inhibitory effect of PGE2 upon the phosphorylation of DNA-PKcs on T2609 in A549 cells (Fig. 4A-C). PKA activation by treating with 6-Phe-cAMP, a PKA-specific activator, increased the phosphorylation at T2609 in SK-MEL-28 cells and HeLa cells but inhibited it in A549 cells, in a similar pattern to that with PGE2 treatment (Fig. 4A-C).
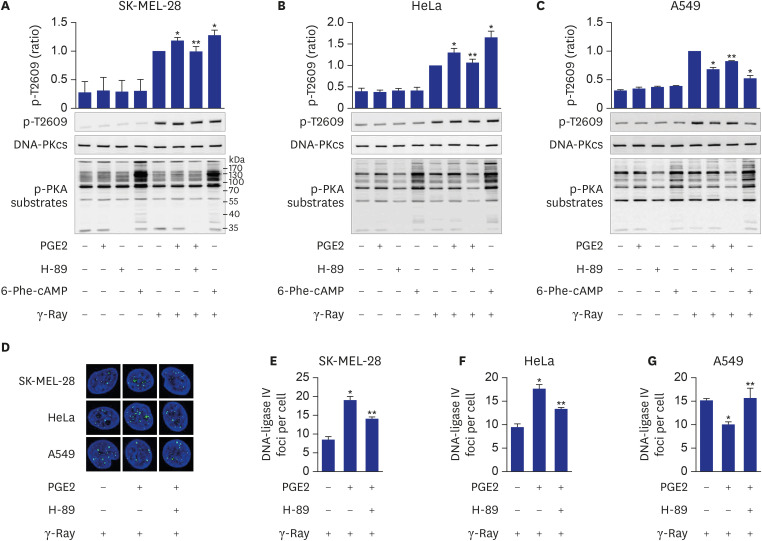
Fig. 4
Mediation of cell-type-specific effects of cAMP by PKA on the phosphorylation of DNA-PKcs at T2609. (A, B, C) Effects of PGE2, H-89 and 6-Phe-cAMP on T2609 phosphorylation resulting from exposure to γ-rays in SK-MEL-28 cells (A), HeLa cells (B), and A549 cells (C). (D) Confocal images of DNA-ligase IV recruited to DSBs following γ-ray irradiation in SK-MEL-28 cells, HeLa cells and A549 cells. (E, F, G) Effects of PGE2 and H-89 on DNA-ligase IV foci per cell, obtained from the images in D of SK-MEL-28 cells (E), HeLa cells (F) and A549 cells (G). The cells were incubated with PGE2 after pretreatment with 20 μM H-89 for 30 minutes or were treated with 50 μM 6-Phe-cAMP for 30 minutes. Next, the cells were exposed to γ-rays, and collected after 1 hour for analysis by western blotting or confocal microscopy. Green color represents XRCC4 and DNA-Ligase IV, and blue color represents DAPI stained nucleus (100-fold amplification). The means ± standard error calculated from the three independent experiments were presented as columns. An asterisk (*) denotes statistically significant differences compared with the irradiated control, and a double asterisk (**) denotes statistically significant differences compared with the PGE2- stimulated cells (P ≤ 0.05, Mann-Whitney U test).
![]()
Inhibition of PKA by H-89 treatment blocked both the stimulatory role of PGE2 upon the recruitment of XRCC4 and DNA-ligase IV to DSB foci in HeLa and SK-MEL-28 cells, and the inhibitory effect of PGE2 upon the recruitment in A549 cells (Fig. 4D-G).
These results indicate that the cell-type-specific modulation of NHEJ repair by cAMP signaling might be mediated by PKA in melanoma cells, uterine cervical cancer cells and lung cancer cells.
DISCUSSION
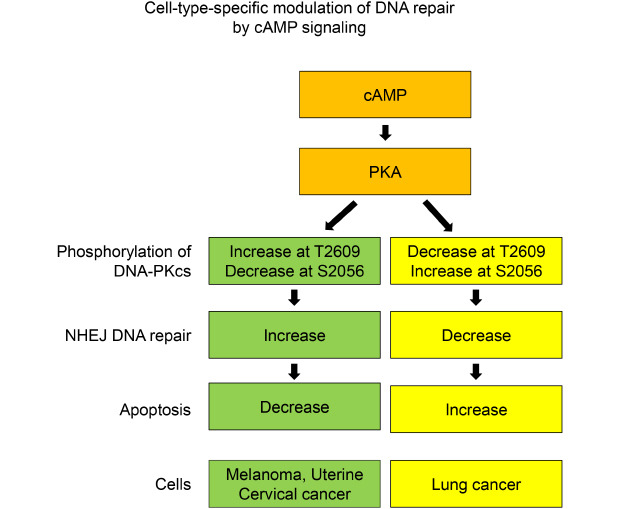
The present study aimed to investigate whether cAMP signaling modulates DNA repair differently depending on the cell type, and we found that cAMP signaling modulates the NHEJ repair of DNA DSBs induced by γ-ray irradiation in malignant melanoma cells, uterine cervical cancer cells, and lung cancer cells and that the cell type specificity might be mediated by PKA-dependent phosphorylation of DNA-PKcs.
cAMP signaling was found to modulate the NHEJ repair of DNA damage resulted from γ-ray irradiation differently depending upon the cell type. Activation of cAMP signaling of malignant melanoma cells and uterine cervical cancer cells by treatment with PGE2 or isoproterenol decreased DNA damage, as assessed by the γ-H2AX assay following γ-ray irradiation, accelerated the disappearance of γ-H2AX, increased NHEJ efficiency, as determined using fluorescent reporter plasmids, stimulated the recruitment of XRCC4 and DNA-ligase IV, major players in NHEJ, to DSB foci, and decreased apoptosis. By contrast, activation of cAMP signaling in lung cancer cells increased DNA DSBs, delayed the disappearance of γ-H2AX, decreased NHEJ efficiency, reduced the recruitment of XRCC4 and DNA-ligase IV, and augmented apoptosis resulting from exposure to γ-rays.
The present study showed that cAMP signaling modulates NHEJ repair differently depending on the cell type and modulates DNA repair similarly in cells of similar origin, such as lung cancer cells (A549 cells and H1299), melanoma cells (SK-MEL-2 and SK-MEL-28) and cervical cancer cells (HeLa and SiHa). This study compared the NHEJ in the different cells following the same treatment with PGE2 and γ-ray irradiation. The cell-type-specific modulation of cAMP signaling of DNA repair seems to result in the cell-type-specific modulation of apoptosis: cAMP signaling stimulated NHEJ repair and inhibited apoptosis in γ-ray-irradiated melanoma cells and cervical cancer cells, whereas cAMP signaling inhibited NHEJ and stimulated apoptosis resulting from γ-ray irradiation in lung cancer cells. Comparable cell-type-specific regulation by cAMP signaling has already been described in cellular proliferation and apoptosis.131415 The cell-type-specific regulation by cAMP signaling is speculated to result from cell-type-specific expression of various proteins that function in cAMP signaling, DNA repair, proliferation, and apoptosis. However, more extensive studies on the modulation of NHEJ and other DNA repair mechanisms by cAMP signaling using various cell types are needed to generalize the cell-type-specific modulation of DNA repair by cAMP signaling.
The roles of cAMP signaling in DNA repair have been reported in melanocytes and melanoma cells. Melanocyte-stimulating hormones (MSHs) bind to and activate melanocortin receptors, which activate cAMP signaling in melanoma cells and melanocytes. Activated cAMP signaling leads to the acceleration of nucleotide excision repair to remove DNA photoproducts caused by UV light independent of pigmentation1617 and cisplatin-induced DNA adducts.18 Our study indicates that cAMP signaling also stimulates the NHEJ repair of radiation-generated DSBs in melanoma cells, a finding similar to the report that MSH prevented the reactive oxygen species-induced increase in γ-H2AX in melanoma cells.19 Thus, cAMP signaling seems to stimulate the nucleotide excision repair of UV-induced DNA damage and NHEJ of γ-ray-induced DNA damage in melanocytes and melanoma cells.
Most uterine cervical cancers are associated with infections of high-risk human papillomavirus (HPV).20 HPV oncogenes, E5, E6 and E7, can stimulate the cyclooxygenase-prostaglandin pathway, which controls inflammation and carcinogenesis, by elevating the expression of cyclooxygenases and cAMP-linked PGE2 receptors, resulting in chronic inflammation and cervical carcinogenesis.212223 Treatment of cervical cancer cells with lactate was reported to enhance the repair of DNA damage resulted from treatment with neocarzinostatin, doxorubicin and cisplatin.24 Because lactate receptor (HCAR1 or G-protein-coupled receptor 81, GPR81), was reported to inhibit adenylate cyclase, cAMP signaling might mediate the lactate effects on DNA repair.25 However, the role of cAMP upon DNA repair in cervical cancer cells remained unclarified. Thus, to our best knowledge, we showed the stimulatory effect of cAMP signaling on the NHEJ repair of DSBs following γ-ray irradiation in cervical cancer cells for the first time. The finding that cAMP inhibits γ-ray-induced apoptosis in cervical cancer cells, together with the report that cAMP signaling inhibits the apoptosis in cervical cancer cells resulting from cisplatin treatment,26 suggests common mechanisms for cAMP signaling to modulate DNA repair and apoptosis following γ-ray radiation and cisplatin treatment in cervical cancer cells.
cAMP signaling was reported to repress base excision repair10 and NHEJ repair11 of DNA damage caused by exposure of lung cancer cells to γ-rays and to augment radiation-generated apoptosis in lung cancer cells.27 These reports completely agree with and support our finding that cAMP signaling modulates the NHEJ repair of DNA damage and apoptosis following γ-ray irradiation.
We also found that the cell-type specificity might be mediated by the PKA-dependent phosphorylation of DNA-PKcs. This finding is derived from the results that cAMP differently modulated the phosphorylation of DNA-PKcs following γ-ray irradiation depending on the cell type, that treatment with the PKA-specific activator induced similar phosphorylation patterns to those with PGE2 treatment and that the inhibition of PKA abolished the PGE2 effects on the phosphorylation of DNA-PKcs and the recruitment of DNA-ligase IV and XRCC4 in all the cell types tested.
DNA-PKcs is phosphorylated at multiple threonine and serine residues by DNA-PK itself and many other protein kinases including ATM.28 The phosphorylation of DNA-PKcs in the ABCDE region containing T2609 and in the PQR region containing S2056 is suggested to affect DNA-PK activity and end processing of damaged DNA, resulting in the alteration of DNA repair activity.29 Differential phosphorylation of DNA-PKcs recruited to DNA ends functions as a molecular switch controlling end-processing and end ligation in NHEJ.30 cAMP signaling was reported to modulate NHEJ repair and differential phosphorylation of DNA-PKcs on S2056 and T2609 lung cancer cells exposed to γ-rays.11 This study showed that cAMP signaling modulates the differential phosphorylation of DNA-PKcs at S2056 and T2609 differently depending on the cell type (cervical cancer cells, lung cancer cells, and melanoma cells), likely resulting in the differential modulation of NHEJ repair activity depending upon the cell type. This study also indicated that PKA mediates the cell-type-specific modulation by cAMP of the differential phosphorylation of DNA-PKcs on S2056 and T2609 following exposure to γ-rays. cAMP signaling also inhibited T2609 phosphorylation in PP2A- and ATM-dependent pathways in lung cancer cells as reported previously11 and increased T2609 phosphorylation in PP2A-independent pathways in cervical cancer cells and melanoma cells.
In summary, the present study showed that cAMP signaling modulates NHEJ repair in cell-type-specific manners following γ-ray irradiation and that PKA mediates the differential phosphorylation of DNA-PKcs at T2609 and S2056 depending on the cell type. In melanoma and uterine cervical cancer cells, PKA activated by cAMP increases the phosphorylation of DNA-PKcs on T2609 with a concomitant decrease in S2056 phosphorylation, which might enhance DNA-PK activity to stimulate NHEJ repair. By contrast, in lung cancer cells, PKA decreases the phosphorylation of DNA-PKcs on T2609 with a concomitant increase in S2056 phosphorylation, which might inhibit DNA-PK activity and delay NHEJ repair.
The finding that cAMP signaling modulates the repair of radiation-induced DNA damage in a cell type- and tissue-specific manner suggests that activity of cAMP signaling might affect the repair of DNA damage caused by radiotherapy to treat in cancer patients. Therefore, therapeutic efficiency of radiotherapy could be improved by modulating the activity of cAMP signaling to inhibit DNA repair of cancer cells or to stimulate DNA repair of normal tissues surrounding metastatic cancer cells.
In conclusion, cAMP signaling modulates the cell-type-specific NHEJ repair of γ-ray-induced DNA DSBs in melanoma cells, uterine cervical cancer cells and lung cancer cells, and these effects may be mediated by PKA-dependent, cell-type-specific differential phosphorylation of DNA-PKcs. This study suggests that the cell type- and tissue-specific modulation of DNA damage repair by cAMP signaling might be applied to improve the therapeutic efficiency of radiation therapy for cancer.
 XML Download
XML Download