

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Apolipoprotein E (apoE) plays a significant role in the normal catabolism of triglyceride-rich lipoprotein constituents [1]. A defect in lipoprotein metabolism in apoE-knockout (KO) mice results in hypercholesterolemia and atherosclerosis [2]. The apoE-KO mouse model develops extensive lesions associated with all phases of atherosclerosis throughout the arterial tree [3]. Accordingly, apoE-KO mice are used as representative models of atherosclerosis [24]. Several cellular and molecular events in the vessels are involved in the development of atherosclerosis [5]. Atherosclerosis is a thickening or hardening of the arteries that is caused by a gradual deposit of atheromatous plaques in the inner lining of an artery [67]. Atheromatous plaques are composed of sticky lipid substances, cellular waste products, and calcium [7]. Atherosclerotic progression to thrombosis involves smooth muscle cell proliferation, cell death and plaque rupture [89]. High-fat diet and high-cholesterol diet are commonly used to induce atherogenic status in atherogenesis-prone animals. Long-term fat-feeding of apoE-KO mice is a useful model of atherosclerotic plaque rupture [10]. Currently, the apoE-KO mouse model has been used for the development of new drugs to protect against atherosclerosis [11].
Atherosclerosis is a chronic inflammatory and complicated vessel disease [812]. Evidence suggests that immune cells, including monocytes and macrophages, are recruited to the subendothelial space of vessels and contribute to the development of atherosclerotic plaques together with extracellular matrix components [513]. Infiltrated monocytes differentiate into macrophages in the subendothelial intima, and induce inflammation and subsequent foam cell formation by taking up modified low-density lipoprotein (LDL) via scavenger receptors (SR), thereby promoting cholesterol loading [814]. Lipid-laden macrophages play a key role in atherosclerotic plaque rupture by producing various proinflammatory mediators and coagulants, as well as reactive oxygen species [8]. It has been shown that the inflammatory reactions of macrophages can influence the development of atherosclerotic plaques [15]. Accordingly, numerous studies have developed therapeutic strategies targeting inflammation and immunity in atherosclerosis [1617]. Currently, treatment for arteriosclerosis is mainly focused on reducing risk factors such as hyperlipidemia. Several anti-inflammatory strategies have emerged as potential treatments for atherosclerotic disease, in addition to existing lipid-lowering therapies [18]. However, treatments targeting the inflammatory disposition associated with atherosclerosis are still limited.
Polyphenols have attracted much attention due to their health-promoting effects, including their antioxidant, anti-inflammatory, antimicrobial and neuroprotective properties, making them valuable in biomedical applications [1920]. Ellagic acid (Fig. 1A) is a polyphenolic compound present in berries, pomegranates and nuts that has antioxidant, anti-inflammatory and antiproliferative properties [2122]. Our previous study showed that ellagic acid inhibited atherosclerosis by promoting cholesterol efflux from lipid-laden foam cells [23]. Based on previous results showing the in vitro effects of ellagic acid on atherogenesis, the present study investigated the atheroprotective effects of ellagic acid on inflammation-associated atherosclerotic lesion formation in vivo. It was examined whether ellagic acid reduced atherosclerotic lesions in atherogenesis-prone animals fed a cholesterol-rich diet through ameliorating vascular inflammation. This study examined various inflammatory mediators and atherosclerotic plaques in the aorta vasculature of apoE-KO mice. In addition, systemic inflammation was examined by measuring plasma levels of proinflammatory mediators. The current results established that dietary ellagic acid may be an atheroprotective agent that reduces vascular inflammation and lessens atherosclerotic lesion formation.
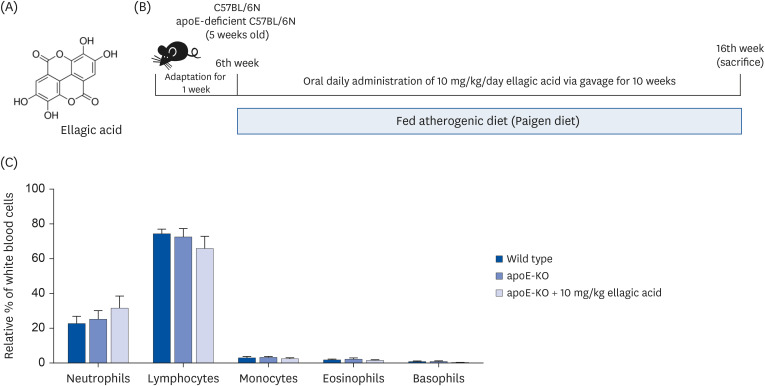
Fig. 1
Wild-type mice and apoE-deficient (apoE-KO) mice were fed an atherogenic Paigen diet for 10 weeks daily with and without oral administration of 10 mg/kg ellagic acid. Cells in blood were counted using a Hemavet HV950 Multispecies Hematologic Analyzer (Drew Scientific). (A) Chemical structure of ellagic acid, (B) schematic illustration of animal experimental design/timeline, and (C) leukocyte profile in blood.
apoE, apolipoprotein E; KO, knockout.
MATERIALS AND METHODS
Chemicals
Ellagic acid and other chemicals were supplied by Sigma Chemical (St. Louis, MO, USA), as were all other reagents unless specifically stated otherwise. Fetal bovine serum (FBS) was obtained from BioWhittaker (San Diego, CA, USA). Antibodies for CD68, vascular cell adhesion molecule-1 (VCAM-1), intracellular adhesion molecule-1 (ICAM-1), P-selectin, platelet endothelial cell adhesion molecule-1 (PECAM-1), nitric oxide synthase 2 (NOS2), heme oxygenase-1 (HO-1) and tissue factor (TF) were supplied by Santa Cruz Biotechnology (Santa Cruz, CA, USA). Monocyte chemoattractant protein-1 (MCP-1) antibody were obtained from Abcam (Waltham, MA, USA). Antibodies for ATP-binding cassette transporter (ABC) A1, ABCG1 and SR-B1 were purchased from Novus Biologicals (Littleton, CO, USA). Horseradish peroxidase (HRP, Rockland Immunochemicals, Pottstown, PA, USA)-conjugated goat anti-rabbit immunoglobulin G (IgG), goat anti-mouse and donkey anti-goat IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). β-Actin antibody was obtained from Sigma Chemicals (St. Louis, MO, USA).
Animals and diets
Wild type C57BL/6 mice (male, 5 weeks old, average body weight of 20 g) and homozygous apoE-KO mice (C57BL/6 background) were provided by Shizuoka Prefecture Laboratory Center (Hamamatsu, Japan). Mice were individually housed in cages and maintained kept on a 12-h light and dark cycle at 23 ± 1ºC with 45–65% relative humidity under specific pathogen-free conditions. The mice were allowed to acclimatize for a week before commencing the experiments. All mouse experiments were performed in accordance with the University’s Guidelines for the Care and Use of Laboratory Animals approved by the Committee on Animal Experimentation of Hallym University (Hallym2011-10).
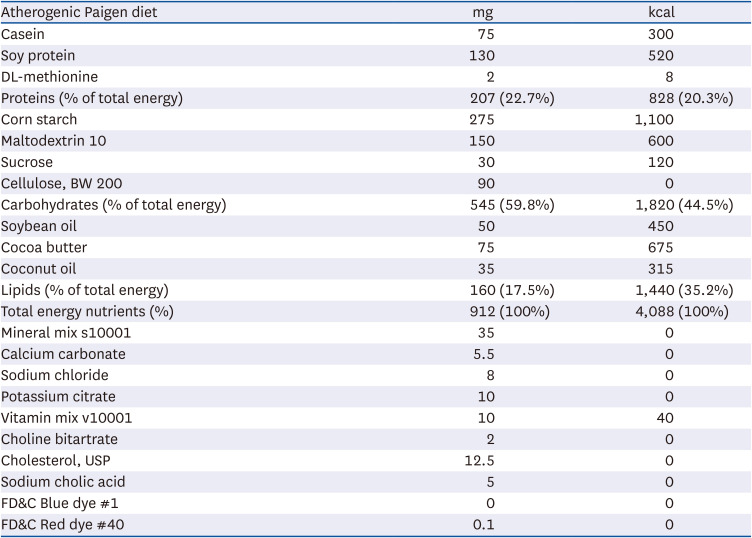
All mice were fed a Paigen diet (40 kcal% fat, 1.25% cholesterol, 0.5% sodium cholate [D12336; Research Diets, Inc., New Brunswick, NJ, USA]) for 10 weeks. The diet constituents were shown in Table 1. The atherogenic Paigen diet is known to develop severe atherosclerotic plaques [2425]. Animal experimental design and timeline were depicted in Fig. 1B. Mice were allocated to 3 groups; 1) wild type fed Paigen diet, 2) apoE-KO mice fed Paigen diet, 3) apoE-KO mice fed Paigen diet and given 10 mg/kg ellagic acid as gavage. After 10 weeks of the diet intervention, mice were sacrificed under zoletin/lumphoon anesthesia. Blood was collected from the abdominal aorta into ethylenediaminetetraacetic acid-coated tubes and plasma was obtained by centrifugation at 3,000 rpm for 10 min and stored at −70°C. The aortas were collected, frozen in liquid nitrogen, and kept at −80°C until used for Western blotting, or were preserved and fixed in 4% paraformaldehyde for immunohistochemical analyses.
Table 1
Composition of atherogenic Paigen diet
The numbers of leukocytes in blood were determined using a Hemavet HV950 Multispecies Hematologic Analyzer (Drew Scientific, Oxford, CT, USA). ApoE deficiency did not alter the leukocyte profile in the blood of mice fed Paigen diet (Fig. 1C). In addition, ellagic acid did not change the amounts of neutrophils and lymphocytes in blood.
Assessment of atherosclerotic lesions
Atherosclerotic lesions were assessed by measuring lipid deposition in the mouse aorta. After the mice were euthanized, the aortas were dissected with removal of adventitial fat, fixed with 4% paraformaldehyde overnight and soaked with 30% sucrose for dehydration. Whole aorta were opened longitudinally from the aortic arch to the iliac bifurcation, mounted en face, and stained lipids with 0.5% Sudan IV dissolved in acetone:ethanol:double distilled water (50:35:15) for 15 min. Overstained colors were reduced by 80% ethanol, and then rinsed with tap water for 1 h. Images for atherosclerotic plaque areas were captured using an optical microscope AXIOIMAGER (Zeiss, Göttingen, Germany). To determine lipids accumulation in the atherosclerotic lesions, cut-aorta tissues were stained with pre-warmed oil red O solution for 10 min in 60°C oven. After staining with 85% propylene glycol for 5 min, stained tissues were observed with Zeiss microscopes.
Staining with hematoxylin and eosin (H&E)
For histologic H&E staining for atherosclerotic lesions, aorta tissues were fixed and dehydrated with 4% paraformaldehyde and 30% sucrose. Tissues were embedded with Tissue-Tek OCT compound and cut by a cryostat microtome (Leica Microsystems, Nussloch, Germany) into 5–6 μm thickness. After staining with Mayer’s hematoxylin for 30 s, the tissues were further stained with alcoholic eosin Y solution for 15 s. Following the dehydration steps from 95% ethanol to 100% ethanol, the stained tissues were observed with Zeiss microscopes. In addition, the diameter of blood vessels and the size of atherosclerotic lesions stained with the H&E staining were quantitatively assessed using Zeiss software.
Enzyme-linked immunosorbent assay (ELISA)
Plasma levels of C-reactive protein (CRP), soluble VCAM-1 (sVCAM-1), tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ) and MCP-1 were measured by using ELISA kits (R&D Systems, Minneapolis, MN, USA). All the procedures were followed according to the manufacturer’s instructions. After reacting plasma samples on microplate wells pre-coated with a biotin-conjugated antibody, avidin conjugated to HRP was added to plate wells. 3,3,5,5-Tetramethylbenzidine (R&D systems) substrate was added to wells for detecting color change, and the enzyme-substrate reaction was terminated by the addition of 3 N sulfuric acid. The developed colors on plate wells were measured at λ = 450 nm.
Western blot analysis
After homogenizing and isolating proteins from the aorta tissues, proteins were loaded on sodium dodecyl sulphate-polyacrylamide gels (SDS-PAGE). Western blot analysis was conducted using whole aorta tissue extracts. Whole tissue extracts were prepared in a lysis buffer containing 1 M β-glycerophosphate, 10% SDS, 0.5 M NaF, 0.1 M Na3VO4 and protease inhibitor cocktail. Tissue extracts containing equal amounts of total proteins were electrophoresed on 6–12% SDS-PAGE and transferred onto a nitrocellulose membrane. Nonspecific binding was blocked by soaking the membrane in a Tris buffered saline-Tween 20 (TBS-T) buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.1% Tween 20) supplemented 5% skim milk or 3% bovine serum albumin for 3 h. the membrane was incubated overnight at 4°C with polyclonal rabbit antibodies of VCAM-1, ICAM-1, P-selectin, NOS2, HO-1, TF, ABCA1, ABCG1 or SR-B1. After three washes with TBS-T, the membrane was incubated for 1 h with goat anti-rabbit IgG or rabbit anti-mouse IgG conjugated to HRP. The individual protein level was determined using immobilon Western chemiluminescent HRP substrate (Merck Millipore, Billerica, MA, USA) and Agfa X-ray film (Agfa-Gevaert, Mortsel, Belgium). Incubation with monoclonal mouse β-actin antibody was also performed for comparative controls.
Immunohistochemical staining
The OCT compound-embedded aorta tissues were cut at 5–6 um thickness, and were fixed in cold acetone (−20°C) for 10 min and rehydrated with phosphate-buffered saline for 5 min. To enhance antigen-antibody affinity, tissue slides were permeabilized with 0.1% Triton X-100 for 10 min. To prevent non-specific antibody binding, the tissue slides were incubated in 10% FBS for 1 h at room temperature. A specific primary antibody against CD68, MCP-1, VCAM-1 and PECAM-1 was incubated overnight with the sectioned tissues. Subsequently, the tissue sections were incubated for 1 h with Cy3-conjugated anti-mouse IgG or fluorescein isothiocyanate (FITC)-conjugated anti-mouse IgG. For identification of nuclei, the fluorescent nucleic acid dye of 4′,6-diamidino-2-phenylindole (DAPI) was applied for 10 min. In addition, VCAM-1 were visualized with 3,3′-diaminobenzidine (DAB) to produce a brown staining, being counterstained with hematoxylin. Images of the stained tissues on slides were obtained with an optical microscope Axioimager system equipped for fluorescence illumination (Zeiss). In addition, the fluorescence intensity was quantified by using ImageJ software.
Statistical analysis
The data are presented as means ± standard error of the mean (SEM). Statistical analyses were conducted by employing Statistical Analysis Systems statistical software package (IBM SPSS Statistics Version 25.0; IBM Corp., Armonk, NY, USA). Significance was determined by one-way analysis of variance, Dunnett’s test or Duncan range test for multiple comparisons. Differences were considered significant at P < 0.05.
RESULTS
Inhibition of aorta plaque lesion formation by ellagic acid
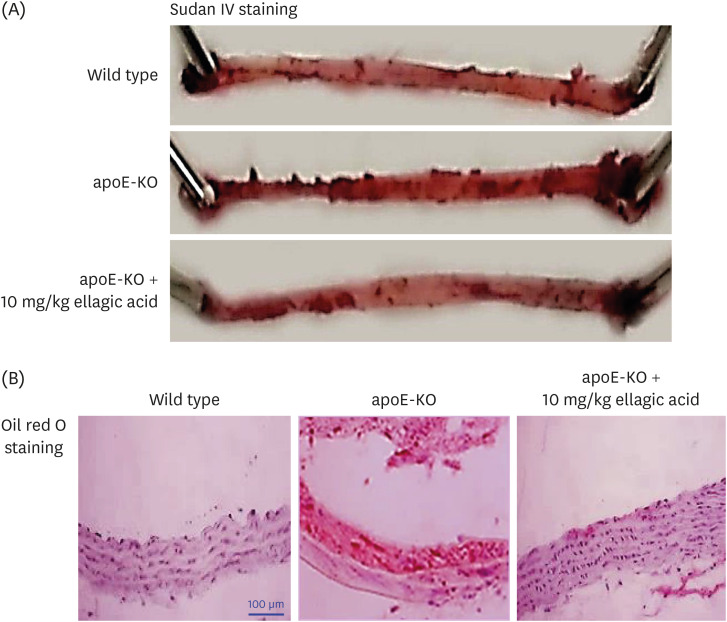
This study examined whether ellagic acid inhibited formation of lipid-laden atherosclerotic plaque lesions, as evidenced by Sudan IV staining of mouse aortas. Compared to wild type mice, the en face lesion area in apoE-KO mice was highly enhanced, especially in the aorta arch (Fig. 2A). However, oral administration of 10 mg/kg ellagic acid reduced plaque lesion formation in the aorta trees. In addition, the increase in the aorta lipid deposition in Paigen diet-fed apoE-KO mice was markedly diminished by ellagic acid (Fig. 2B).
Fig. 2
Inhibition of lipid accumulation in aorta by ellagic acid. After apoE-deficient (apoE-KO) mice were orally treated with 10 mg/kg ellagic acid for 10 weeks, aortas were extracted. Aortas of wild type C57BL/6 mice and untreated apoE-KO mice were also obtained. Aorta was longitudinally opened from the aorta arch to the iliac bifurcation. Aorta was fixed with 4% buffered formalin overnight and dehydrated with 30% sucrose. (A) To determine whether lipid-enriched plaques were formed, Sudan IV staining was performed within whole aortas. For lipid accumulation in aorta, oil red O staining was done with aorta tissues cut in 5 μm thickness. (B) Counter-staining was conducted with hematoxylin. Magnification: ×200 (n = 6, each of groups). Scale bar = 100 μm.
apoE, apolipoprotein E; KO, knockout.
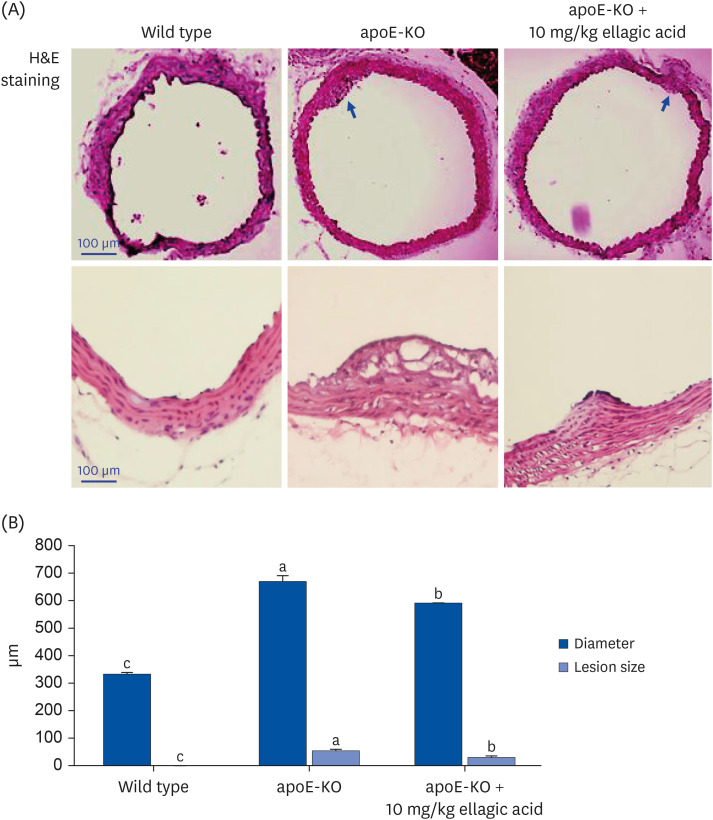
Histological examination of the aortas with H&E staining was performed and the size of the atherosclerotic lesions quantified with an image analysis system. The H&E staining of the aorta cross-sections showed that the apoE-KO mice had larger aorta lesion size, compared to wild type mice (Fig. 3A). However, there was a significant reduction in lesion size in ellagic acid-supplied apoE-KO mice (Fig. 3B). Accordingly, ellagic acid diminished aorta lesions increased by apoE deficiency.
Fig. 3
Effects of ellagic acid on atherosclerotic lesion formation within aortas. After apoE-deficient (apoE-KO) mice were orally treated with 10 mg/kg ellagic acid for 10 weeks, aortas were extracted. Aortas of wild type C57BL/6 mice and untreated apoE-KO mice were also obtained. Aorta was longitudinally opened from the aorta arch to the iliac bifurcation and was fixed with 4% buffered formalin overnight and dehydrated with 30% sucrose. (A) To examine atherosclerotic lesion formation on aorta wall, H&E staining was performed with aorta tissues cut in 5–6 μm thickness. Stained tissues were observed by microscopy with 200× magnification (n = 6, each of groups). (B) The extent of atherosclerotic areas was expressed as lesion diameter and size in the entire aortic surface area. Scale bar = 100 μm.
apoE, apolipoprotein E; KO, knockout; H&E, hematoxylin and eosin.
a-cValues in bar graphs not sharing a same lower case indicate significant different at P < 0.05.
Suppression of systemic inflammation by ellagic acid
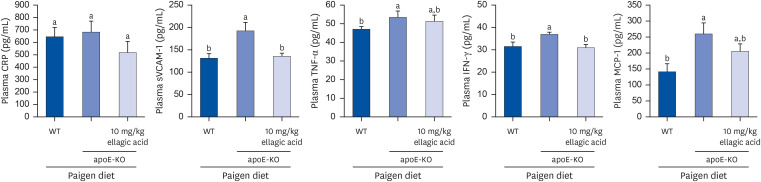
The current study investigated that apoE deficiency induced vascular inflammation in cholesterol-fed mice, which was attenuated by oral administration of ellagic acid. Plasma levels of the inflammatory mediators of CRP, sVCAM-1, TNF-α, INF-γ and MCP-1 were measured with ELISA kits, following the indicated treatments. Plasma levels of inflammatory mediators other than stress-related CRP were significantly higher in apoE-KO mice than in wild type mice (Fig. 4). However, plasma levels of sVCAM-1 and INF-γ were strongly reduced in apoE-KO mice treated with 10 mg/kg ellagic acid for 10 weeks. Plasma levels of CRP, TNF-α, and MCP-1 tended to decline in ellagic acid-treated apoE-KO mice (Fig. 4). Accordingly, ellagic acid may alleviate systemic inflammation in apoE-KO mice.
Fig. 4
Effects of ellagic acid on systemic and vascular inflammation. After apoE-deficient (apoE-KO) mice were orally treated with 10 mg/kg ellagic acid for 10 weeks, blood was collected and centrifugated at 3,000 rpm for 10 min to obtain plasma. Blood samples of wild type C57BL/6 mice and untreated apoE-KO mice were also collected. Plasma levels of CRP, sVCAM-1, TNF-α, IFN-γ, and MCP-1 were measured by using ELISA kits. All the ELISA procedures were followed according to the manufacturer’s instructions.
apoE, apolipoprotein E; KO, knockout; CRP, C-reactive protein; sVCAM-1, soluble vascular cell adhesion molecule-1; TNF-α, tumor necrosis factor-α; IFN-γ, interferon-γ; MCP-1, monocyte chemoattractant protein-1; ELISA, enzyme-linked immunosorbent assay; SEM, standard error of the mean; WT, wild type.
a,bData (mean ± SEM, n = 3–6) in respective bar graphs without a common letter differ at P < 0.05.
Modulation of aorta vessel inflammation by ellagic acid
Aorta sections were immunohistochemically stained with green FITC-conjugated CD68 antibody and red Cy3-conjugated MCP-1 antibody against macrophages. Compared to that of wild type mice, the expression of the macrophage biomarkers of CD68 and MCP-1 in aorta vessels were greatly increased in apoE-KO mice that fed cholesterol (Fig. 5A). In contrast, the induction of fluorescent CD68 and MCP-1 was noticeably reduced in apoE-KO mice treated with ellagic acid. These results suggest that ellagic acid can inhibit inflammation-associated atherosclerosis in apoE-KO mice.
Fig. 5
Effects of ellagic acid on induction of induction of inflammatory biomarkers in aortas. After apoE-deficient (apoE-KO) mice were orally treated with 10 mg/kg ellagic acid for 10 weeks, aortas were extracted (n = 6, each of groups). Aortas of wild type C57BL/6 mice and untreated apoE-deficient mice were also obtained. (A) For the measurement of protein induction of CD68 and MCP-1 in aorta vessels, the CD68 localization was identified as FITC-green staining and the MCP-1 was visualized with Cy3-red staining. Nuclear staining was done with DAPI (blue). Each photograph is representative of 3–6 mice. Magnification: 200-fold. Scale bar = 100 μm. Total tissue extracts were prepared for Western blot analysis with a primary antibody against (B) NOS2, (C) HO-1, and (D) TF. The bar graphs (mean ± SEM, n = 3) represent quantitative results of the upper bands obtained from a densitometer. β-Actin protein was used as an internal control.
apoE, apolipoprotein E; KO, knockout; MCP-1, monocyte chemoattractant protein-1; FITC, fluorescein isothiocyanate; DAPI, 4′,6-diamidino-2-phenylindole; NOS2, nitric oxide synthase-2; HO-1, heme oxygenase-1; TF, tissue factor; SEM, standard error of the mean; WT, wild type.
a-cValues in bar graphs not sharing a same lower case indicate significant different at P < 0.05.
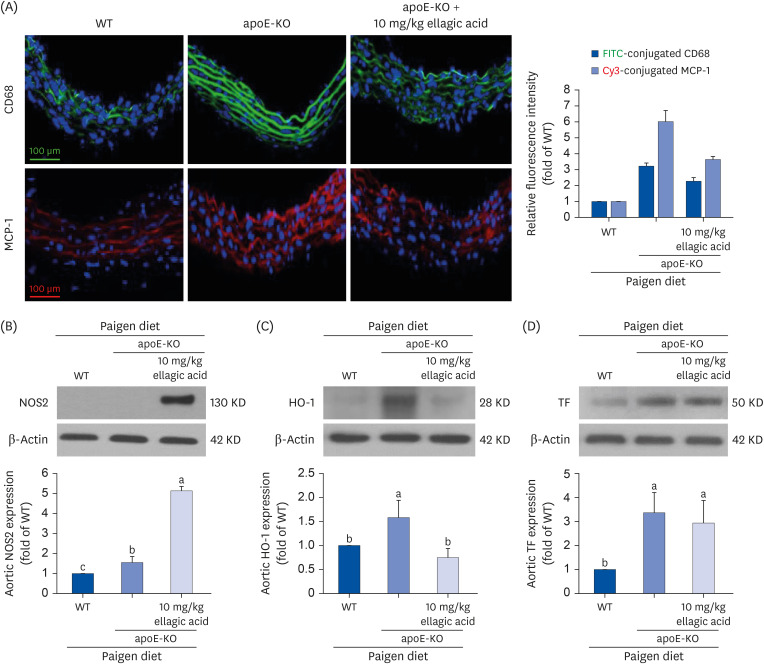
Other studies have reported that inducible NOS deficiency reduces atherosclerosis in apoE-KO mice [2627]. Western blot analysis was performed to determine whether apoE deficiency promoted NOS2 induction in the aorta, and this effect was attenuated by treating apoE-KO mice exposed to a high cholesterol-Paigen diet with 10 mg/kg ellagic acid. Aorta level of NOS2 was significantly increased in apoE-KO mice, and was further enhanced by supplementation with ellagic acid (Fig. 5B). On the other hand, apoE deficiency highly induced aorta expression of HO-1 and TF in Paigen diet-fed apoE-KO mice (Fig. 5C and D). When 10 mg/kg ellagic acid was administered to these mice, the aorta HO-1 expression was significantly reduced. No significant reduction was observed in the TF expression of ellagic acid-treated apoE-KO mouse aorta (Fig. 5D).
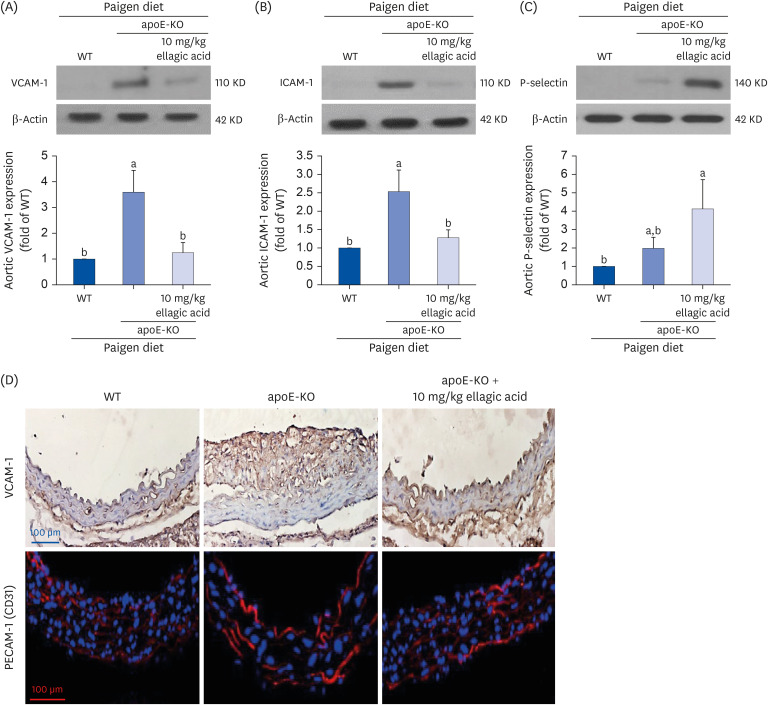
Inhibitory effects of ellagic acid on aorta induction of adhesion molecules
Vascular endothelial adhesion molecules including VCAM-1 and ICAM-1 are overexpressed in the initial stage of atherosclerosis, which may lead to upregulation of atherosclerotic lesions [2829]. Western blot data showed that the cell adhesion molecules of VCAM-1 and ICAM-1 were highly induced in apoE-KO mice (Fig. 6A-C). When apoE-KO mice fed a Paigen diet were treated with 10 mg/kg ellagic acid, levels of these adhesion molecules in aorta tissues were reduced (Fig. 6A and B). In contrast, the treatment of ellagic acid tended to further increase P-selectin level (Fig. 6C). On the other hand, an immunohistochemical staining was conducted to examine tissue levels of VCAM-1 and PECAM-1. As shown in Fig. 6D, apoE-KO mice have increased aorta expression of brown DAB-visualized VCAM-1 and red fluorescent Cy3-colored PECAM-1, whereas the levels of VCAM-1 and PECAM-1 decreased in the aortas of ellagic acid-treated apoE-KO mice.
Fig. 6
Blockade of induction of cell adhesion molecules by oral treatment of ellagic acid. After apoE-deficient (apoE-KO) mice were orally treated with 10 mg/kg ellagic acid for 10 weeks, aortas were extracted (n = 6, each of groups). Aortas of wild type C57BL/6 mice and untreated apoE-deficient mice were also obtained. Tissue extracts were prepared for Western blot analysis with a primary antibody against (A) VCAM-1, (B) ICAM-1, and (C) P-selectin. The bar graphs (mean ± SEM, n = 3) represent quantitative results of the upper bands obtained from a densitometer. β-Actin protein was used as an internal control. (D) Aorta tissue level of VCAM-1 was determined by visualizing with brown DAB and being counterstained with hematoxylin. In addition, PECAM-1 was immunohistologically stained with red Cy3-conjugated second antibody. Counterstaining was performed with blue DAPI. Each photograph is representative of 3–6 mice. Magnification: 200-fold. Scale bar = 100 μm.
apoE, apolipoprotein E; KO, knockout; VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intracellular adhesion molecule-1; SEM, standard error of the mean; DAB, 3,3′-diaminobenzidine; PECAM-1, platelet endothelial cell adhesion molecule-1; DAPI, 4′,6-diamidino-2-phenylindole; WT, wild type.
a,bValues in bar graphs not sharing a same lower case indicate significant different at P < 0.05.
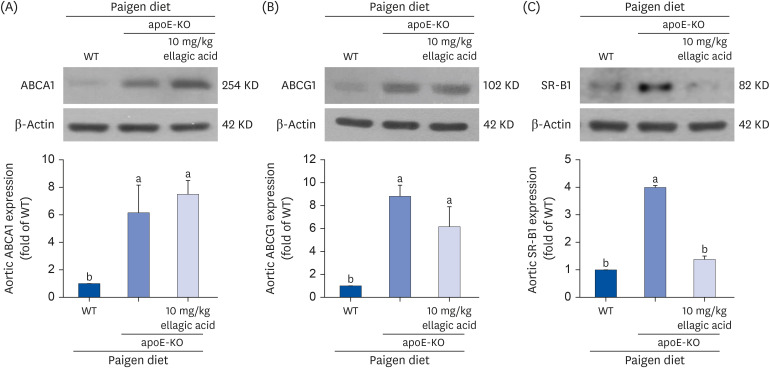
Modulation of cholesterol handling by ellagic acid
One study revealed that in the presence of apoE, ABCA1 overexpression modulated high density lipoprotein and apoB-containing lipoprotein metabolism and reduced aorta atherosclerosis [30]. Compared to those of wild type mouse aortas, the levels of the cholesterol efflux-related transporters of ABCA1 and ABCG1 were markedly increased in apoE-KO mouse aortas (Fig. 7A and B). Oral administration of ellagic acid did not affect the levels of these transporters. On the other hand, the apoE deficiency elevated aorta level of SR-B1, a scavenging receptor of modified LDL, of cholesterol-fed mice (Fig. 7C). In contrast, oral administration of 10 mg/kg ellagic acid inhibited the SR-B1 induction in the aortas of apoE-KO mice (Fig. 7C).
Fig. 7
Effects of ellagic acid on cholesterol trafficking in aortas. After apoE-deficient (apoE-KO) mice were orally treated with 10 mg/kg ellagic acid for 10 weeks, aortas were extracted (n = 6, each of groups). Aortas of wild type C57BL/6 mice and untreated apoE-deficient mice were also obtained. Tissue extracts were prepared for Western blot analysis with a primary antibody against (A) ABCA1, (B) ABCG1, and (C) SR-B1. The bar graphs (mean ± SEM, n = 3) represent quantitative results of the upper bands obtained from a densitometer. β-Actin protein was used as an internal control.
apoE, apolipoprotein E; KO, knockout; ABC, ATP-binding cassette transporter; SR-B1, scavenger receptor class type B1; SEM, standard error of the mean; WT, wild type.
a,bValues in bar graphs not sharing a same lower case indicate significant different at P < 0.05.
DISCUSSION
This study was conducted to explore the effects of ellagic acid on vascular inflammation and atherosclerotic plaque formation in apoE-KO mice. Our major findings indicate that ellagic acid supplementation significantly reduces atherosclerotic plaque lesion formation and lipid deposition in the aorta, lowers levels of inflammatory mediators such as sVCAM-1 and IFN-γ, and modulates the expression of key proteins involved in inflammation, including CD68, MCP-1, VCAM-1, ICAM-1, and PECAM-1, as well as proteins associated with plaque stability, such as aortic NOS2 and HO-1. These results suggest that ellagic acid can attenuate apoE deficiency-induced vascular inflammation and atherosclerotic plaque formation.
It is known that apoE deficiency elevates plasma cholesterol due to impaired clearance of cholesterol-enriched lipoproteins [12]. Additionally, plasma accumulation of cholesterol-rich lipoproteins results in the development of severe hypercholesterolemia and spontaneous atherosclerotic lesions in apoE-deficient mice, similar to what has been observed in humans [310]. Thus, the apoE-deficient mouse model is well established for the study of human atherosclerosis. This study revealed gross plaque lesions in cholesterol-fed apoE-KO mice, compared to cholesterol-fed wild type mice. Consistently, lipid deposition was clearly observed in the aorta tree of apoE-KO mice, which is an atherosclerotic model. Accordingly, the current animal experimental design/timeline is thought to be well structured. Currently, the apoE-KO mouse model is used to develop new drugs against atherosclerosis to test compounds for the treatment of the risk factors related to hypercholesterolemia, hypertension, and inflammation [1131]. Interestingly, ellagic acid reduced the plaque lesion area and lipid deposition in the aorta tree of apoE-KO mice.
Atherosclerosis is a chronic inflammatory disease, that is initiated by hypercholesterolemia in most cases [781631]. In addition, apoE protects against atherosclerosis through the direct regulation of chronic inflammatory responses and inhibition of T cell proliferation [32]. Atherosclerosis-prone animals such as apoE-deficient mice have many immune system components within atherosclerotic lesions [3233]. This study showed that apoE deficiency did not influence the plasma leukocyte profile, compared to that of cholesterol-fed wild type mice. However, apoE-KO mice exhibited increased plasma levels of the proinflammatory sVCAM-1, IFN-γ and TNF-α, which were derived from T cells and macrophages. IFN-γ primes macrophages to produce chemokines and cytotoxic molecules, induces the expression of the genes encoding adhesion molecules on endothelial cells, and regulates lipid uptake [34]. In addition, cholesterol-fed apoE-KO mice exhibited increased expression of the inflammatory cytokines of CD68 and MCP-1 in macrophages and monocytes and activation of the adhesion molecules of VCAM-1 and PECAM-1 in the aorta. Accordingly, vascular inflammation occurred in cholesterol-fed apoE-KO mice with gross plaque lesions. These inflammatory mediators have emerged as therapeutic targets for inflammation and immunity in atherosclerosis [17].
Numerous studies have shown that anti-inflammatory compounds inhibit the formation of atherosclerotic plaques [173536]. However, the problem is that adverse immunosuppressive effects are associated with the systemic use of anti-inflammatory drugs. Natural anti-inflammatory products and their polyphenols and have attracted much attention, mainly due to their safety and therapeutic potential in biomedical applications and their accessibility through daily food intake [192037]. A previous study revealed that purple perilla frutescens extracts containing α-asarone inhibited inflammatory atheroma formation in dyslipidemic apoE-deficient mice [38]. Polyphenolic ellagic acid, which has anti-inflammatory properties, accelerates cholesterol efflux from lipid-laden foam cells [23]. In fact, our previous study showed that the spleen wet weight increased in ellagic acid-administered apoE-KO mice, indicating that ellagic acid may improve inflammatory responses [39]. The current study demonstrated that ellagic acid ameliorated atherosclerotic plaque lesions in cholesterol-fed apoE-KO mice by possibly alleviating systemic inflammation derived from T cells, macrophages and endothelial cells. In addition, ellagic acid strongly inhibited the activation of macrophages and endothelial cells in the aorta of cholesterol-defective mice. Accordingly, one can assume that the inhibition of vascular inflammation by ellagic acid was responsible for reducing atherosclerotic plaques in cholesterol-fed apoE-KO mice.
The dietary polyphenols of quercetin and theaflavin attenuate atherosclerosis in apoE gene-KO mice by alleviating inflammation, increasing NO bioavailability, and inducing HO-1 [37]. The present study showed that ellagic acid greatly induced aorta NOS2 in apoE-KO mice, suggesting that this compound may improve NO bioavailability. Surprisingly, this study showed that apoE deficiency increased the aorta tissue levels of HO-1 and TF in cholesterol-fed mice, and ellagic acid highly reduced the HO-1 level without significant change in the TF level. HO-1, an inducible enzyme responsible for the breakdown of heme, is primarily considered an antioxidant and a target for treatment of chronic inflammatory diseases [40]. On the other hand, oxidative stress can induce HO-1 under pathological conditions [4142]. Induction of cellular HO-1 is a signature of oxidative stress for its downstream effects under pro-oxidative states [42]. It is thought that oxidative stress may result in cholesterol-fed apoE-KO mice, leading to upregulation of aorta HO-1 level that was inhibited by treatment of ellagic acid.
Foam cell formation and lesion development are known to be caused by passage of LDL cholesterol into the artery wall and its engulfment by macrophages [43]. SR-B1 in endothelial cells mediates the delivery of LDL into arteries and promotes its accumulation by artery wall macrophages, thereby stimulating atherosclerosis [44]. In our previous study [23], ellagic acid diminished SR-B1 induction and elevated ABCA1 induction in lipid-laden macrophages, thereby improving cholesterol efflux and blocking foam cell formation. Similarly, oral administration of ellagic acid inhibited the SR-B1 induction in the aortas of apoE-KO mice and tended to further increase ABCA1 induction. Ellagic acid reduced the macrophage biomarkers of CD68 and MCP-1 in aorta vessels of cholesterol-fed apoE-KO mice, indicating its inhibition of accumulation of inflammatory macrophages carrying SR-B1 in the aorta wall. One study shows that the SR-B1 expression increases in atherosclerosis-prone regions of the mouse aorta before lesion formation, and in human atherosclerotic arteries [45]. Inhibition of the endothelial delivery of LDL into artery walls by SR-B1 may be a new therapeutic target in the battle against atherosclerosis.
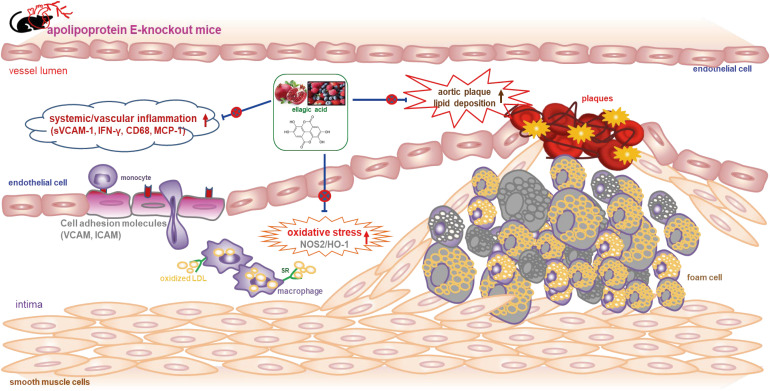
In summary, the current report demonstrated that ellagic acid inhibited atherosclerotic plaque formation and lipid deposition in the aorta tree of cholesterol-rich Paigen diet-fed apoE-KO mice (Fig. 8). In addition, ellagic acid diminished systemic inflammation in apoE-KO mice by reducing the plasma levels of sVCAM-1 and the T cell cytokines of TNF-α and IFN-γ. Oral administration of ellagic acid suppressed the induction of macrophage-expressing MCP-1 and CD68 and endothelial cell-expressing adhesion molecules of VCAM-1 and PECAM-1 in cholesterol-fed apoE-KO mice. In addition, ellagic acid attenuated vascular cholesterol uptake by inhibiting the aorta SR-B1, although there was no significant change in cholesterol efflux by ABCA1 or ABCG1. Therefore, ellagic acid may lessen inflammation-associated atherosclerotic plaques by acting as an anti-inflammatory agent in atherosclerosis-prone apoE-KO mice. These findings are of considerable significance, as they suggest a wide range of effects on endothelial function and atherosclerosis, as well as the anti-inflammatory properties of ellagic acid. Thus, ellagic acid may be an alternative to atheroprotective apoE during episodes of hypercholesterolemia. Although there is good evidence that ellagic acid exerts antiatherogenic and anti-inflammatory effects on animal models, the evidence in humans is limited.
Fig. 8
Diagram illustrating effects of ellagic acid on atherosclerosis in apoE-deficient (apoE-KO) mice. Ellagic acid reduced atherosclerosis by alleviating aortic plaque formation and lipid deposition. Furthermore, ellagic acid diminished vascular inflammation by down-regulating expression of sVCAM-1, IFN-γ, CD68, and MCP-1.
apoE, apolipoprotein E; KO, knockout; sVCAM-1, soluble vascular cell adhesion molecule-1; IFN-γ, interferon-γ; MCP-1, monocyte chemoattractant protein-1; VCAM, vascular cell adhesion molecule; ICAM, intracellular adhesion molecule; NOS2, nitric oxide synthase 2; HO-1, heme oxygenase-1; LDL, low-density lipoprotein; SR, scavenger receptor.
 XML Download
XML Download