

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Ischemic heart disease (IHD) has been recognized as a primary cause of death in patients with cardiovascular disorders; its pathophysiology is the inability of the left ventricular (LV) myocardium to pump the required blood flow to meet metabolic demands [1]. Despite both pharmacological and interventional advances, the current therapeutic strategy for IHD provides limited prevention of heart failure after the occurrence of IHD. This is especially true for patients who have been treated with reperfusion within a short period following IHD and have experienced extensive LV myocardial damage subsequent to advanced heart failure and even cardiovascular death [2]. More effective therapeutic options have long been required to protect and improve damaged LV myocardial function after the occurrence of IHD. Furthermore, recent research has shown that when oxygen and substrates required for respiration are lacking due to ischemia, the mitochondrial tricarboxylic acid cycle and oxidative phosphorylation are interrupted and the mitochondrial membrane voltage collapses, causing oxidative damage and apoptosis [3-5]. Ischemia-reperfusion (IR) leads to Ca2+ overload and the formation of reactive oxygen species, which open the mitochondrial permeability transition pore, release cytochrome c into the cytoplasm, and activate caspase-3 [6].
Normal mitochondrial function supplies the energy necessary for cell survival through the production of adenosine triphosphate (ATP), which is abundant in the liver, brain, heart, kidney, and muscle cells [7,8]. In particular, mitochondria occupy more than 30% of the volume of cardiomyocytes, provide 95% of the ATP required for heartbeat, and serve as metabolic hubs for oxidative phosphorylation, the citric acid cycle, and fatty acid β-oxidation [9]. Cyclosporine, MTP-131, and TRO4030 are used in the pharmacological treatment of mitochondrial damage, inhibiting mitochondrial membrane permeability and preventing cell death. However, the precise mechanism underlying this phenomenon is not yet known [6]. Another option may be mitochondrial transplantation, which involves the transfer of mitochondria into LV myocardial cells. Mitochondrial transplantation is a method of separating mitochondria from externally derived tissues or cells and transplanting them into new tissues or cells [10]. When mitochondria enter IR-damaged myocardial cells through transplantation, energy production is increased by replenishing mitochondrial DNA (mtDNA), and the cells are restored to their proper function [10,11]. To date, methods for transplanting mitochondria in vitro have included co-incubation, direct microinjection, cell-penetrating peptides, mitoception, photothermal nanoblade use, magnetomitotransfer, Mitopunch, Fluid FM, and mitochondrial delivery of the extracellular vesicles, among others [10]. However, these methods have limitations in reflecting in vivo situations because they represent phenomena that occur in a test tube. In addition, although research on direct mitochondrial transfer was conducted before cell-mediated mitochondrial transfer, its mechanism and strategy remain controversial. Therefore, we aimed to provide preliminary results for clinical therapeutic applications by directly transplanting mitochondria into LV myocardial cells using the Langendorff system.
The Langendorff system is an experimental method developed by Oscar Langendorff in 1895 that allows the heart to beat in vitro by performing retrograde perfusion of an isolated heart [12]. When an insertion tube is connected to the aorta and a solution containing oxygen and nutrients is perfused, the solution is perfused into the coronary artery by the aortic valve, supplying oxygen and nutrients to the myocardium, and the perfusate is discharged into the right atrium. By obstructing the oxygen supply to the myocardium, conditions such as ischemia, myocardial infarction, and reperfusion injury can be reproduced and studied [13].
In the present study, mitochondrial transplantation was performed using the Langendorff system after creating a situation similar to that of IR in vitro. We aimed to investigate the therapeutic feasibility of mitochondrial transplantation as a treatment for IHD via the following experiments. First, the rat heart was connected to the Langendorff system to create IR and post-IR mitochondrial transplantation states. Second, after the Langendorff perfusion experiment, the oxygen consumption capacity of the mitochondria in the myocardial tissue was measured, and the respiratory control rate (RCR) was calculated to confirm the ATP production capacity of the mitochondria. Third, the number of mitochondrial copies was measured using real-time PCR. The ATP production capacity per mitochondrial unit was confirmed by the second and third experiments. Fourth, to identify candidate genes related to IHD, gene expression levels were measured using real-time PCR.
METHODS
Laboratory animals
Sprague–Dawley rats (male, 7 weeks old, body weight 200 ± 10 g) (Coatech) were used for all experiments. Animal testing was approved by the Chung-Ang University Animal Experiment Ethics Committee (approval number: 202301020113). The breeding environment was maintained at a temperature of 22°C ± 3°C, humidity of 50% ± 10%, ventilation of 14 to 18 times/h, lighting of 150 to 300 lx, and light/dark cycle of 12 h. Water and food were freely consumed during the experimental period. Twelve animals were randomly assigned to three groups: the normal group (CON, control group), ischemia-reperfusion group (IR), and ischemia-reperfusion-mitochondrial transplantation group (IR + transpl). Three animals in each group were utilized for assessing mitochondrial function, and the fourth was used for Evans staining. All animal experiments were performed in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines, the Laboratory Animal Act, and animal testing regulations.
Heart isolation
The animals were anesthetized by intravenous injection (1 ml/kg) of alfaxalone (5 mg/kg, Alfaxan; JUROX Pty Limited), xylazine hydrochloride (5 mg/kg, Rompun inj.; BAYER KOREA Ltd.), and heparin (300 IU/kg, Greencross Heparin Sodium Inj.; GC Biopharma Corporation) [14,15]. After confirming the absence of the flexion reflex, the rats were euthanized via heart excision. The aorta of each isolated rat heart and the cannula were then securely ligated with sutures. Subsequently, each heart was installed onto the Langendorff system and retrogradely perfused with Normal Tyrode’s (NT) solution. This solution contained 143 mM NaCl, 5.4 mM KCl, 5 mM HEPES, 0.5 mM MgCl2, 0.3 mM NaH2PO4, 1.8 mM CaCl2 and 5.5 mM glucose, and was adjusted to pH 7.4.
Animal cardiac ischemic model
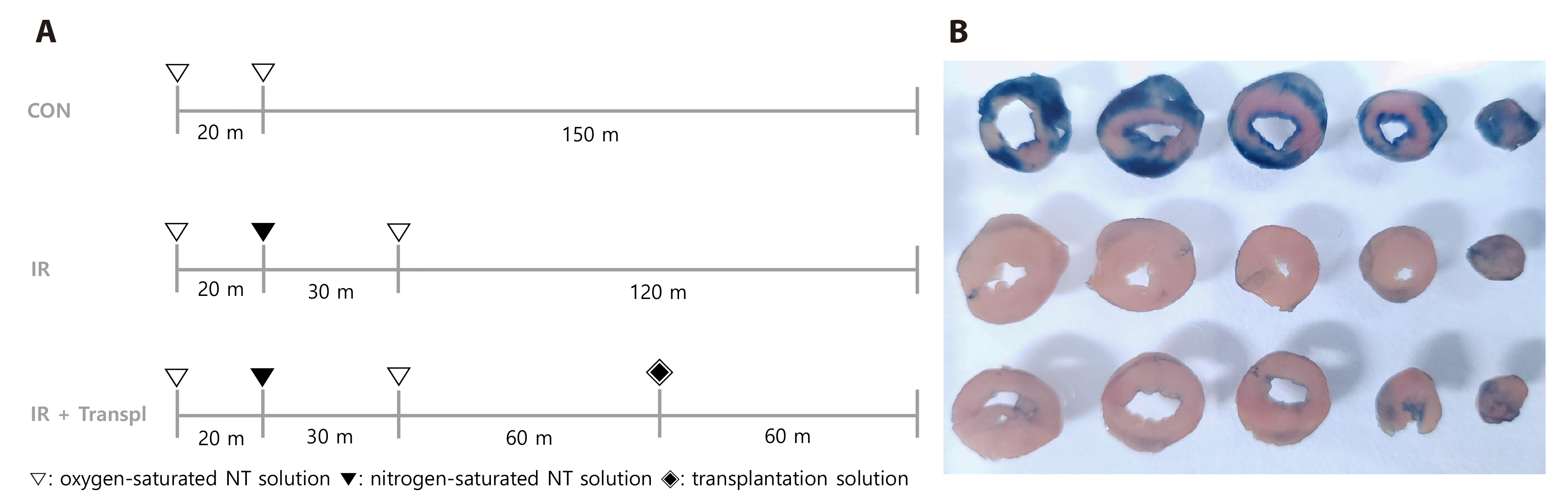
The animal cardiac ischemic model was modified as previously described [15]. The Langendorff systems were washed using flowing sterilized water and NT solution for 5 min each, and the experiment was performed after preheating the Langendorff systems to 37°C using a constant temperature circulating water tank (Changshin Science). All of the isolated hearts were stabilized via perfusion with oxygen-saturated NT solution for 20 min. The control group was perfused with an oxygen-saturated NT solution for 150 min, and the IR group was perfused with a nitrogen-saturated NT solution for 30 min to induce ischemia and then perfused with an oxygen-saturated NT solution for 60 min to induce reperfusion injury. In the IR + transpl group, IR damage was induced in the same manner as in the IR group, and the isolated mitochondria were perfused for 60 min, followed by transplantation (see Fig. 1A). The heart was perfused retrogradely in constant flow mode, and the flow rate was adjusted to 7 ± 1 ml/min. The heart rate (HR) was measured via video observation lasting for 10 sec.
Isolation and transplantation of mitochondria
After euthanasia, approximately 1 g of the gluteus maximus muscle was collected, transferred to a 50 ml tube, and mixed with 5–10 ml mitochondrial isolation buffer (MIB) 1 solution (180 mM KCl, 0.5 mM EDTA-Na2, 10 mM Tris-base, pH 7.4 at 4°C). The muscle was then cut into small pieces with medical scissors and then homogenized with an overhead stirrer. The homogenate was centrifuged at 1,000 g for 10 min at 4°C, the supernatant was transferred to a new tube, and centrifugation (1,000 g, 10 min, 4°C) was repeated. Subsequently, the supernatant was decanted to a new tube and centrifuged (7,000 g, 10 min, 4°C), and the resulting supernatant was removed. The pellet was resuspended in 5–10 ml of MIB 2 solution (180 mM KCl, 0.5 mM EDTA-Na2, 10 mM Tris-base, 1 g/l BSA, pH 7.4 at 4°C) and centrifuged (7,000 g, 10 min, 4°C). The resulting supernatant was removed, resuspended with 5–10 ml of MIB 2 solution, centrifuged (7,000 g, 10 min, 4°C) once more, and the supernatant was removed to obtain a mitochondrial pellet. For quality control, the mitochondria content was validated through RCR, and quantification was based on protein levels using the Bradford assay. The transplantation solution was prepared with 230 ng of mitochondrial protein per 200 ml of NT solution and perfused for 60 min [16].
Confirmation of the non-infarcted area
After completing the experimental protocol, 0.6 ml of 0.25% Evans blue dye was uniformly infused into the aortic cannula orifice. Subsequently, the heart was frozen at −20°C for 2 h and sliced into 5 mm thick transverse sections. The presence of a blue stain demarcated the non-ischemic area; conversely, a pale negative stain indicated the infarcted myocardium and/or the viable myocardium in the area at risk [17,18].
Preparation of permeabilized fibers from heart tissue
The details of our experiment have been previously described by Kuznetsov et al. [19]. The LV endocardium of the perfused heart was immersed in isolation solution A (10 mM Ca-EGTA solution, 0.1 μM Ca, 20 mM imidazole, 20 mM taurine, 49 mM K-MES, 3 mM K2HPO4, 9.5 mM MgCl2, 5.7 mM ATP, 15 mM phosphocreatine, 1 μM leupeptin, pH 7.1) containing 10 mM Ca-EGTA solution (2.77 mM CaK2EGTA stock solution, 7.23 mM K2EGTA stock solution) (CaK2EGTA stock solution: 100 mM CaCO3, 100 mM EGTA, 200 mM KOH, K2EGTA stock solution: 100 mM EGTA, 200 mM KOH). Then, 50 μg/ml saponin was added to 2 ml of the stripped tissue and vortexed (minimum speed, 20 min, 4°C) to create permeable tissue. Afterwards, to remove saponin and ATP, respiration medium B (0.5 mM EGTA, 3 mM MgCl2·6H2O, 20 mM taurine, 10 mM KH2PO4, 20 mM HEPES, 1 g/l BSA, 60 mM K-lactobionate, 110 mM mannitol, 0.3 mM DTT, pH 7.1) (60 mM K-lactobionate: 500 mM K-lactobionate stock solution) and 1 mg/ml fatty acid-free BSA was added to the tissue and vortexed (minimum speed, 5 min, 4°C). The procedure was repeated three to four times, followed by an experiment measuring the oxygen consumption capacity of the mitochondria.
Measurement of mitochondrial oxygen consumption capacity
Tissues and solutions were placed inside a chamber (Instech) connected to a Neofox system (Ocean Optics) to measure oxygen consumption capacity. The oxygen consumption rate (OCR) was measured using the NeoFox viewer (version 2.30; Ocean Optics) program. After the chamber was preheated to 37°C using a constant temperature circulating water tank (Changshin Science), 100 μl of respiration medium B solution was added into the chamber, and the magnetic stir bar was operated for 10–15 min until the viewer's graph was stabilized. Subsequently, permeable tissue, G/M (10 mM glutamate + 5 mM malate), and 2 mM adenosine diphosphate were added (in that order), and oxygen consumption capacity was measured by observing the change in oxygen inside the chamber. After the slopes of states 3 and 4 had been calculated, the RCR was calculated as follows [20-22] (Supplementary Fig. 1):
Mitochondrial copy number (mtDNA-CN) measurement
The mtDNA-CN was measured using endo-myocardial DNA. DNA was obtained using a QIAamp DNA Mini Kit (Qiagen), and 8 ng was used for quantification using a UV spectrophotometer (Nanodrop 1000; Thermo Fisher Scientific). Real-time PCR was performed using a LightCycler 2.0 instrument (Roche Diagnostics). After the cycle of initial denaturation at 95°C for 10 min, denaturation at 95°C for 10 sec, annealing at 60°C for 10 sec, and extension at 72°C for 10 sec, the melting curve was performed 35 times under conditions of 65°C–95°C and 0.1°C/sec. The primers used in this study are listed in Table 1 [23,24]. Because the extracted DNA included genomic DNA (gDNA) and mtDNA, the expression level was quantified by comparing the mtDNA-CN and nuclear gene copy number. The formulas used to quantify the expression level and the RCR per unit of mitochondria were as follows [25]:
Real-time quantitative PCR
Real-time PCR was performed to measure gene expression using a LightCycler 2.0 (Roche Diagnostics). After initial denaturation at 95°C for 10 min followed by 40 cycles of denaturation at 95°C for 10 sec, annealing for 10 sec at an appropriate temperature for each primer, and extension for 10 sec at 72°C, the melting curve was measured at 65°C–95°C and 0.1°C/sec. The primers and denaturation temperatures are listed in Table 1 [23,24]. The expression level of the candidate gene was normalized to the expression level of Gapdh, a reference gene, and the relative quantity was quantified using the 2–ΔΔCT method [26].
Statistical analysis
Values were expressed as mean ± standard error of the mean. The Kruskal–Wallis test was selected because RCR does not require the groups to be normally distributed. Analysis of variance (ANOVA) was utilized to compare mtDNA-CNs and gene expression levels. We employed repeated-measures ANOVA to analyze the time-dependent effects of HR. The Bonferroni test was used to adjust for multiple comparisons among the three groups in the post-hoc analysis. Statistical analyses were conducted using R version 4.3.2 [27], with a significance threshold set at 0.05.
RESULTS
Acute IR injury influenced HR in perfused rat heart
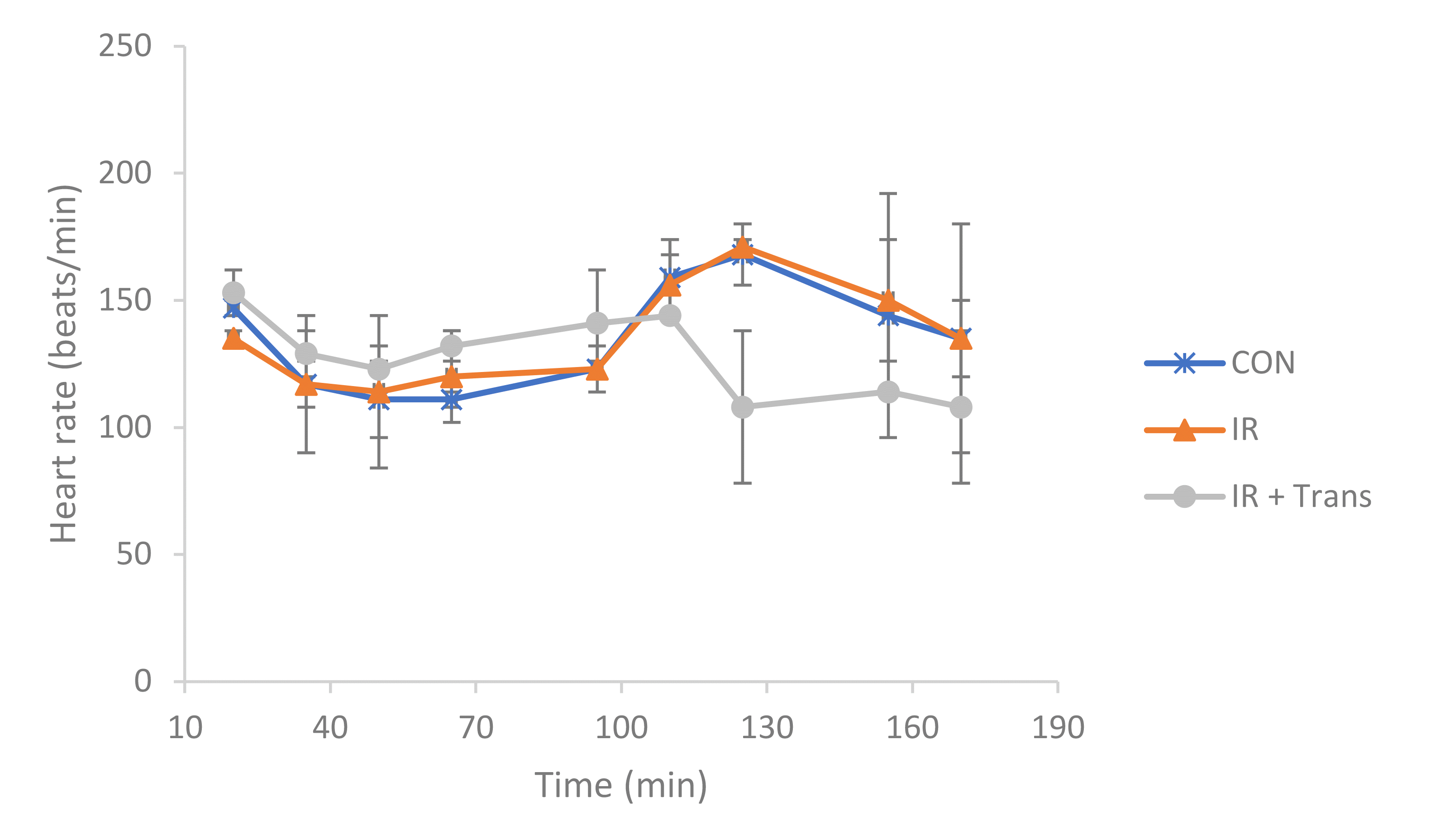
After stabilization, the isolated hearts exhibited a normal beating rate of 145 ± 4 beats/min. Following mitochondrial transplantation, the IR + transpl group displayed a tendency toward a decreased HR compared to the other groups. However, we observed inconsistencies in this result, as all isolated hearts maintained a HR within the normal range [28] seen ex vivo from stabilization to the termination of the experiment, and no statistically significant differences were detected (Fig. 1).
Mitochondrial transplantation did not induce the structural recovery of IR-damaged hearts
We observed areas of the heart that were stained blue (indicating perfused regions), as well as those that did not stain (indicating injured regions). The results revealed that the non-ischemic area in the IR group was distinctly smaller than that in the control group; this was not restored in the IR + transpl group (Fig. 2).
Mitochondrial transplantation revealed a tendency to restore mitochondrial function in IR-damaged hearts
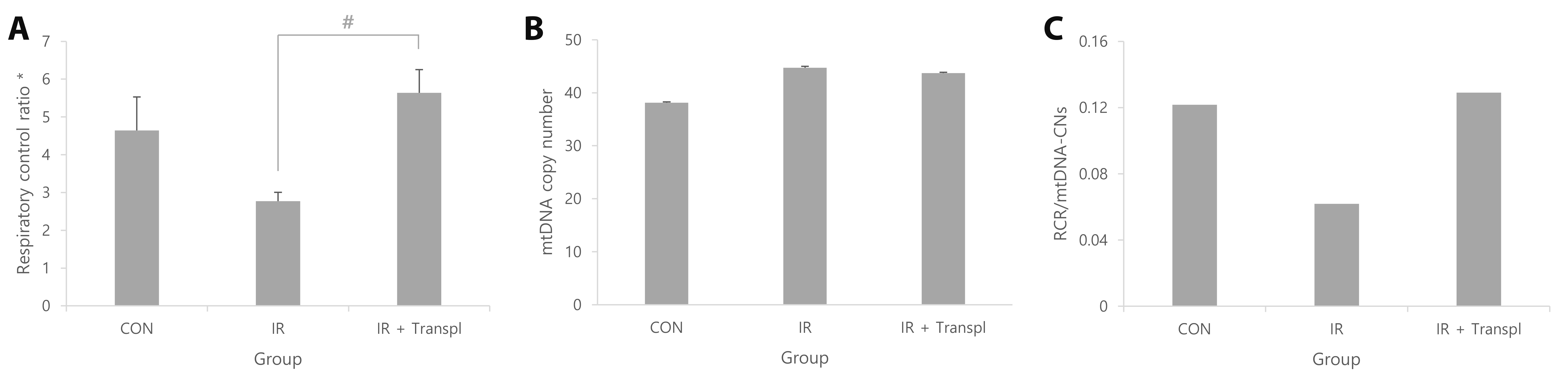
To evaluate mitochondrial function, the oxygen consumption capacity was measured twice for each individual, and the RCR was calculated. Compared with the control group, the IR group showed a tendency toward decreased oxygen consumption capacity (1.68 times lower). Conversely, compared with the IR group, the IR + transpl group showed a tendency toward increased oxygen consumption capacity (2.04 times higher), although this difference did not reach statistical significance (Fig. 3A). mtDNA-CN was observed to be similar in all groups (Fig. 3B). Notably, IR injury led to a decreased RCR to mtDNA-CN ratio, then recovered after mitochondrial transplantation (Fig. 3C).
Mitochondrial transplantation regulated several fatty acid metabolism-related genes
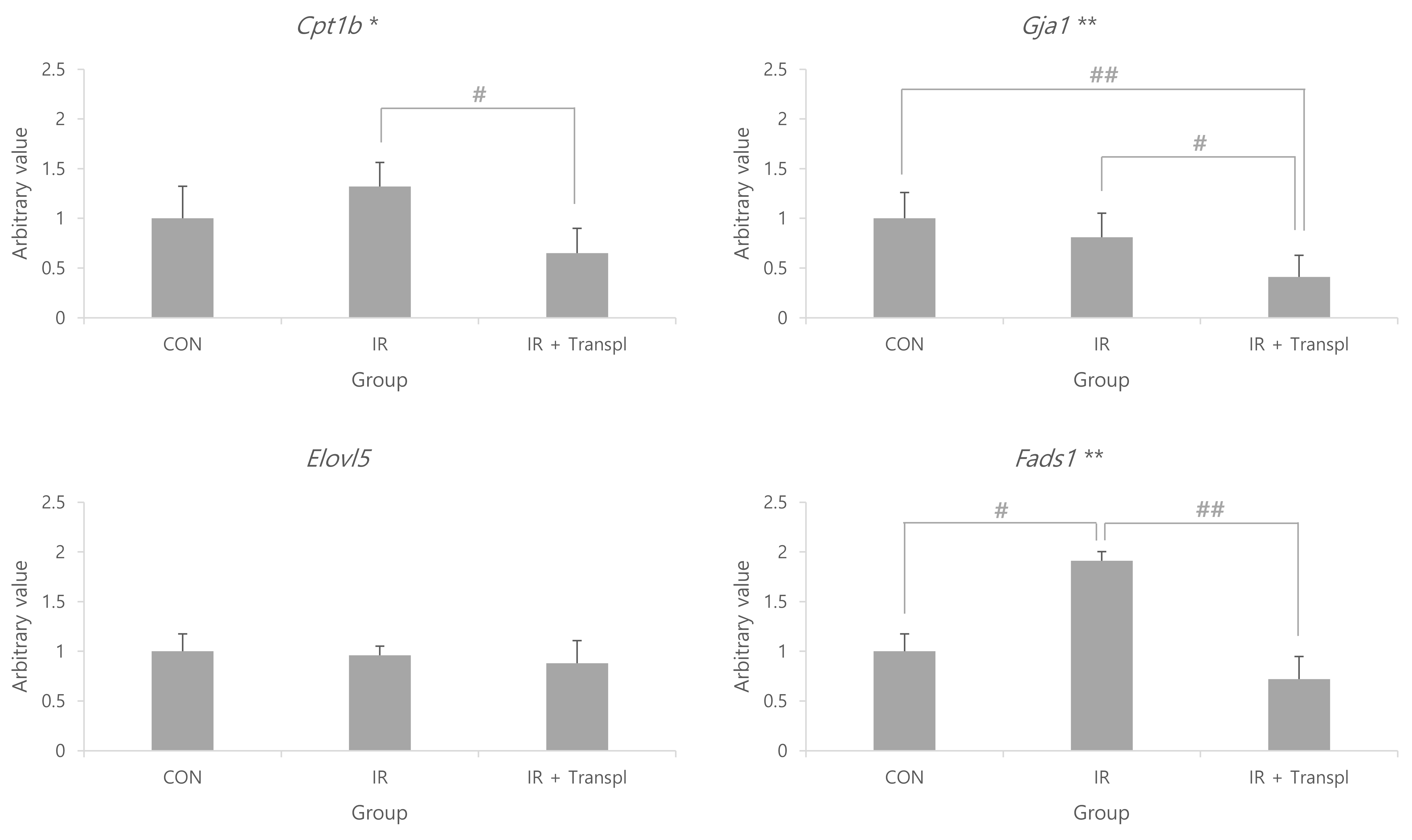
A list of genes linked to fatty energy metabolism in rat skeletal and myocardial tissues, derived from our prior studies [15,23,29], was integrated with the relevant KEGG dataset [rno01212: Fatty acid metabolism - Rattus norvegicus (rat)] [30]. In total, 62 genes were identified, from which we randomly selected four. The relative expression levels of Cpt1b (p < 0.05), Gja1 (p < 0.01), and Fads1 (p < 0.01) differed between the groups, although that of Elovl5 did not. The expression level of Fads1 was higher in the IR group than in the control group (p < 0.05), and was lower in the IR + transpl group than in the IR group (p < 0.01). The expression level of Gja1 was lower in the IR + transpl group than in the control (p < 0.01) or IR groups (p < 0.05) (Fig. 4).
DISCUSSION
There is a dearth of effective therapeutic options for IHD due to rising medical costs and cardiovascular-related mortality rates. Thus, in this study, we sought to determine whether damaged myocardial function could be restored by transplanting mitochondria into the heart under ex vivo IR states. It was previously reported that transplanted mitochondria can remain viable in the myocardium for 28 days although isolated mitochondrial function rapidly decreases within 1 h owing to external factors [31]. Isolated mitochondria from L6 cells were co-incubated with H9C2 cells to further determine the time dependence of mitochondrial transplantation efficiency. As no significant differences were observed within 3 h (data not shown), mitochondrial transplantation was performed for more than an hour in the Langendorff experiment.
The isolated hearts were connected to the Langendorff system, and an IR-damaged state and post-damage mitochondrial transplantation conditions were reproduced. As a result, the HR tended to slow during ischemic conditions compared to that under normoxic condtions, but tended to increase again during reperfusion and mitochondrial transplantation (Fig. 2). It has been reported that under ischemic conditions, the HR increases as a compensation for the lack of oxygen and nutrients being delivered to the myocardium [21,32,33]; however, in this experiment, this was predicted to be the result of the heart failing to interact with surrounding tissues under ex vivo conditions. The simple HR was a poor biomarker in our study because it remained within the normal biological range.
While our findings showed that oxygen consumption capacity was lower in the IR group than in the control group, the oxygen consumption capacity of the IR + transpl group was higher than that of the IR group, although it improved compared to that of the IR group, it did not improve sufficiently to match that of the control group (Fig. 1). Cowan et al. [32] reported that mitochondrial transplantation into cardiac cells, such as myocardial cells and fibroblasts, was possible by culturing cardiac cells under oxygen-deprived conditions to create conditions similar to those of ischemia, followed by co-incubating with a solution containing mitochondria. This has been reproduced using the Langendorff system [32,33]. Research has shown that most transplanted mitochondria fuse with existing mitochondria to form a network, and some are hydrolyzed [31]. In other words, IR damage can be induced using the Langendorff system, and the transplantation method of directly perfusing mitochondria into the heart using the Langendorff system appears to protect myocardial tissue from IR damage. Therefore, mitochondrial transplantation may emerge as a therapeutic option for IHD. Currently, the best-known mechanism by which mitochondria move between cells involves endosomes [11,31,34]. Specifically, mitochondria are transplanted into cardiac cells through actin-dependent endocytosis, which increases energy production and replenishes mtDNA to induce proper cell function [11,31,34]. Under normal conditions, the heart uses fatty acids as the main energy substrate, followed by carbohydrates, ketone bodies, and amino acids; however, it is known to dynamically switch substrate availability to ensure efficient cardiac function [35]. Among the three groups in this study, the Cpt1b and Fads1 genes showed the highest expression in the IR group and the lowest expression in the IR + transpl group (Fig. 4). A previous study showed a reduction in fatty acid oxidation with lipid accumulation in Cpt1b knockout mice, demonstrating that this gene is related to fatty acid oxidation [36]. From this, it can be expected that normal fatty acid oxidation did not occur due to IR damage, and that the expression of Cpt1b increased as a compensation. In the IR + transpl group, the expression of Cpt1b was lower, suggesting the restoration of fatty acid metabolism and myocardial protection effects through mitochondrial transplantation. A previous study demonstrated that the Fads1 gene induces apoptosis and increases reactive oxygen species when silenced, indicating that it is a gene related to the survival of healthy cells [37]. In the IR group, it could be expected that the expression of the Fads1 gene increased due to the excessive production of reactive oxygen species in the myocardium and a protective effect against cell damage. In the IR + transpl group, the expression of the Fads1 gene was reduced but was restored to normal by mitochondrial transplantation, and apoptosis-prevention effects were expected. The expression of connexin 43 (encoded by Gja1) increases under ischemic conditions [38]. Existing research has shown that this gene protects the mitochondria by reducing mitochondrial membrane potential, respiration, and reactive oxygen species production [38]. In this study, the expression of Gja1 was found to be lower in the IR + transpl group than in the IR group, which is thought to be due to the protective effect of mitochondrial transplantation rather than Gja1 itself, resulting in relatively lower expression of Gja1 (Fig. 4). Considering the mitochondrial respiratory capacity in myocardial tissue observed in the above results, it appears that cardiomyocyte dysfunction can be restored by replenishing mitochondria damaged by IHD through mitochondrial transplantation; however, it also functions as normal myocardial cells without IR damage and thus, can be interpreted as not recovering. In the future, based on the results of this study, additional interpretation of the functions of the Cpt1b and Fads1 genes, which appear to be expressed as compensatory effects of IR, will be required. In addition, various transplantation methods will be developed by applying mitochondrial transplantation to an in vivo IR model and methods that can restore function to the levels observed in the normal myocardium will be studied.
Given that acute injury often manifests through metabolic alterations rather than immediate physical or structural changes, our study focused on assessing infarct size using Evans blue staining and detecting IHD-related gene markers via PCR. While additional investigations are warranted to address the limitations in the number of repetitions and to elucidate the underlying mechanisms and potential clinical applications, the findings of this study suggest that mitochondrial transplantation may confer protection against early IR injury through the modulation of cardiac energy metabolism. Therefore, mitochondrial transplantation holds promise as a potential therapeutic intervention for IHD.
SUPPLEMENTARY MATERIALS
Supplementary data including one figure can be found with this article online at https://doi.org/10.4196/kjpp.2024.28.3.209
 XML Download
XML Download