

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Atherosclerosis is a major cause of cardiovascular diseases including myocardial infarction and heart stroke [1]. Hyperlipidemia is a leading cause of atherosclerosis, and high circulating level of low-density lipoprotein cholesterol (LDL-C) is considered as an important determinant of atherosclerosis [23]. Hyperlipidemia is mainly attributed to high intake of high-fat high-cholesterol diets [4]. Accordingly, the management of hyperlipidemia has been suggested as a therapeutic strategy for cardiovascular diseases [25]. On the other hand, the activation of reverse cholesterol transport (RCT) is a key strategy to lessen hyperlipidemia in the rodents [678]. RCT is a multi-step process including peripheral cholesterol efflux, which is involved in removing excess cholesterol via lipoproteins from peripheral tissues to the liver [910]. Excess cholesterol from non-hepatic peripheral cells such as macrophages is transferred to the liver [611]. It is assumed that the disruption of cholesterol efflux or RCT promotes atherogenesis.
Macrophages restore cellular cholesterol homeostasis through activating cholesterol efflux to lipid-poor acceptors with the help of ATP-binding cassette (ABC) transporters [1112]. It has been reported that ABC transporter A1 (ABCA1) inhibits atherosclerosis development in animal models [7]. Cholesterol is converted to cholesteryl esters by lecithin-cholesterol acyltransferase (LCAT), followed by transfer to LDL and very LDL (VLDL) via cholesterylester transfer protein (CETP) in exchange for triglycerides (TG) [1213]. Inhibition of this process by CETP inhibitors may enhance HDL cholesterol (HDL-C) and diminish LDL-C and VLDL [14]. Plasma phospholipid transfer protein (PLTP) manipulates cholesterol metabolism through regulating the size of HDL particles [13]. Pharmacological inhibition of the activity of CETP and PLTP may be a new therapy for dyslipidemia through reducing apolipoproteins B (apoB) secretion [15].
Apolipoproteins E (apoE) accepts cholesterol from cells including macrophages in the vessel wall and transports it back to the liver via RCT process [16]. ApoE present in plasma lipoproteins is involved in the clearance of chylomicrons and VLDL remnants by the liver [1718]. ApoE knockout (apoE KO) mice are lack of function of lipoprotein metabolism and are known as representative models of atherosclerosis and hypercholesterolemia [19]. ApoE KO mice have high levels of plasma cholesterol as a result of this impaired clearance of cholesterol-enriched lipoproteins. There is direct evidence for the cooperative contribution of both apoE and apolipoproteins A1 (apoA1) to cholesterol efflux. ApoE-mediated cholesterol efflux depends on cellular or extracellular positioning of apoE of macrophages [16]. Macrophage apoA1 compensate for apoE deficiency and delay the progression of atherosclerotic lesions by stimulating cholesterol efflux and RCT [20].
Ellagic acid is a polyphenol present in the form of ellagitannin in plants of berries, nuts, and pomegranates [2122]. Ellagic acid inhibits oxidized LDL-mediated lectin-like oxidized LDL receptor-1 (LOX-1) expression, generation of reactive oxygen species, and inflammation in human endothelial cells [23]. Our previous study shows that ellagic acid exhibits anti-atherosclerotic activity by promoting cholesterol efflux from oxidized lipid-laden foam cells [24]. Ellagic acid displays anti-atherogenic activity by upregulating hepatocyte expression of paraoxonase 1 (PON1) [25]. Based on diverse evidence that ellagic acid is anti-atherosclerotic, the current study investigated that ellagic acid improve atherosclerosis through activating RCT process in apoE KO mice. Wild type (WT) mice and apoE KO mice were fed high-fat high-cholesterol Paigen diet for 10 weeks to induce hypercholesterolemia and atherosclerosis. The current study determined plasma levels of apoA1, CETP, PLTP, LCAT and PON1 and expression of ABC transporters, scavenger receptor B1 (SR-B1) and LOX-1 in peritoneal macrophages and livers from WT mice and apoE KO mice.
MATERIALS AND METHODS
Chemicals
Fetal bovine serum (FBS), trypsin–ethylenediaminetetraacetic acid (EDTA), and penicillin–streptomycin were obtained from BioWhittaker (San Diego, CA, USA). Dulbecco’s modified eagle’s media (DMEM), ellagic acid and DMEM chemicals were supplied by Sigma Chemicals (St. Louis, MO, USA), as were all other reagents unless specifically stated otherwise. Ellagic acid was dissolved in dimethylsulfoxide for live culture with cells; its final culture concentration was ≤0.1%. Antibodies for ABCA1 and ABCG1 were purchased from Novus biologicals (Littleton, CO, USA). Antibodies of SR-B1 and LOX-1 were obtained from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Horseradish peroxidase (HRP; Rockland Immunochemicals, Philadelphia, PA, USA)-conjugated goat anti-rabbit immunoglobulin G (IgG), goat anti-mouse and donkey anti-goat IgG were obtained from Jackson ImmunoResearch Laboratories (West Grove, PA, USA). β-Actin antibody was obtained from Sigma Chemicals.
Animals and diets
WT C57BL/6N mice (male, 5 weeks old, average body weight [BW] of 20 g) and homozygous apoE KO mice (C57BL/6 background) were obtained from Japan Shizuoka Laboratory Center (Hamamatsu, Shizuoka, Japan). Mice were housed individually in wire-bottomed cages, fed and watered, and maintained kept on a 12-h light and dark cycle at 23 ± 1°C with 60% relative humidity under specific pathogen-free conditions. The mice were allowed to acclimatize for a week before beginning the experiments. All animal experiments were performed in accordance with the University’s Guidelines for the Care and Use of Laboratory Animals approved by the Committee on Animal Experimentation of Hallym University (Hallym2011-10). All mice were fed Paigen diet (15–20% fat, 1.25% cholesterol, 0.5% sodium cholate [D12336, Research Diets, Inc, New Brunswick, NJ, USA]) for 10 weeks. The diet composition of atherogenic diet was shown in Supplementary Table 1. The atherogenic Paigen diet is known to develop severe atherosclerotic plaques. Some of apoE KO mice orally received 10 mg/kg ellagic acid daily during the same experimental periods. Mice were allocated to 3 groups (16 mice of each diet group); WT mice with atherogenic diet, apoE KO mice with atherogenic diet, and finally apoE KO mice with atherogenic diet and ellagic acid.
After 10 weeks of diet feeding, mice were sacrificed under Zoletin/Lumphoon anesthesia. No mice were dead. Blood was collected from the abdominal aorta into EDTA-coated tubes. Plasma was obtained by centrifugation at 3,000 rpm for 10 min and stored at −70°C. The organs were washed with physiological saline by direct injection in the heart left ventricle. Aortas and the livers were collected and immediately frozen in liquid N2 and stored at −20°C until analysis.
Plasma lipid analyses
After 10 weeks of the diet intervention, plasma levels of total cholesterol (TC) and TG, and HDL-cholesterol (HDL-C) were measured using colorimetric enzymatic assays (Asan Pharmaceuticals, Hwasung, Korea) according to the manufacturers’ instructions. LDL-C was determined using the formula, LDL-C = TC – (HDL-C – TG/5). Based on these values, the atherosclerosis index (AI) was obtained by using the formula, AI = (TC – HDL-C)/HDL-C.
Western blot analysis
Western blot analysis was conducted using J774.A1 macrophages, whole liver tissue extracts and mouse peritoneal macrophage lysates. Liver tissues were homogenized for isolating proteins. Macrophage lysates and whole tissue extracts were prepared in a lysis buffer containing 1 M β-glycerophosphate, 10% sodium dodecyl sulphate, 0.5 M NaF, 0.1 M Na3VO4 and protease inhibitor cocktail. Whole cell lysates were prepared in a 1 M Tris-HCl lysis buffer (pH 6.8) containing 1 M β-glycerophosphate, 1% β-mercaptoethanol, 0.5 M NaF, 0.1 M Na3VO4 and protease inhibitor cocktail. Cell lysates and tissue extracts containing equal amounts of total proteins were electrophoresed on 6–12% from, proteins were loaded on sodium dodecyl sulphate-polyacrylamide gels and transferred onto a nitrocellulose membrane. Nonspecific binding was blocked by soaking the membrane in a TBS-T buffer (50 mM Tris-HCl [pH 7.5], 150 mM NaCl, and 0.1% Tween 20) supplemented 5% skim milk or 3% bovine serum albumin for 3 h. The membrane was incubated overnight at 4°C with polyclonal rabbit antibodies of ABCA1, ABCG1, SR-B1, apoE, apoA1, and LOX-1. After three washes with Tris buffered saline-Tween 20, the membrane was incubated for 1 h with a goat anti-rabbit IgG or a rabbit anti-mouse IgG conjugated to HRP (Rockland Immunochemicals). The individual protein level was determined by reacting with immobilon Western chemiluminescent HRP substrate (Merck Millipore, Billerica, MA, USA) and X-ray film (Agfa-Gevaert, Mortsel, Belgium). Incubation with monoclonal mouse β-actin antibody was also performed for comparative controls.
Isolation of mouse peritoneal macrophages
Fresh peritoneal macrophages were harvested from WT mice and apoE KO mice 3 days after receiving an intraperitoneal injection of 3% thioglycollate and plated in DMEM with 10% FBS. After 2 h incubation at 37°C, non-adherent cells were removed by washing with phosphate-buffered saline, and adherent cells consisting of macrophages were used for further experiments.
Reverse transcription polymerase chain reaction (RT-PCR) analyses
Total RNA was isolated from aorta and liver tissues by using a commercially available Trizol reagent kit (Invitrogen, Waltham, MA, USA) and synthesized to cDNA by using a commercial cDNA synthesis kit (Thermo Fisher Scientific, Waltham, MA, USA). Using a Power SYBR Green master mix, the RT-PCR was conducted with mRNA transcripts of ABCA1, ABCG1, SR-B1, apoA1, apoA1 binding protein (AIBP), LDL receptor (LDLR), and ABCG8. The polymerase chain reaction (PCR) condition was 95°C for 10 min for holding, and 40 cycles at 95°C for 15 sec and at 60°C for 1 min. For the measurement of semi-quantitative RT-PCR, the PCR products (10 µL) were thermo-cycled and electrophoresed on 3% agarose gel containing 0.5 mg/mL ethidium bromide, and the bands were visualized using a Vilber-Lourmat UV transilluminator and gel photographs were obtained. For the measurement of real-time PCR, the operation was done with Applied Biosystems 7500 Real-time PCR system instrument (Thermo Fisher Scientific). Glyceraldehyde 3-phosphate dehydrogenase was used as a housekeeping gene to normalize target gene expression. Detailed information for the PCR primers was shown in Supplementary Table 2.
Enzyme-linked immunosorbent assay (ELISA)
Plasma levels of CETP (MyBioSource, San Diego, CA, USA), PLTP (MyBioSource), and LCAT (Cloud-Clone, Katy, TX, USA) were determined by using commercial sandwich-type ELISA kits. In addition, plasma level of PON1 was measured by using an ELISA kit (R&D Systems, Minneapolis, MN, USA). After reacting plasma samples on microplate wells pre-coated with a biotin-conjugated antibody, avidin-conjugated to HRP was added to plates. The substrate 3,3,5,5-tetramethylbenzidine (Sigma-Aldrich Chemicals, St. Louis, MO, USA) was added to wells for detecting color change, and the enzyme-substrate reaction was terminated by adding 3 N sulfuric acid. The color development from blue to yellow in microwells was measured by spectrophotometry at λ = 450 nm.
Liver tissue staining
To detect the hepatic histological changes, liver tissues were fixed and dehydrated with 4% paraformaldehyde and 30% sucrose. Subsequently, tissues were embedded with optimal cutting temperature compound and cut by a cryostat microtome into 6 μm thickness. Liver tissues were stained with Mayer’s hematoxylin-alcoholic eosin Y (H&E) solution for 30 seconds. Following the dehydration steps with 95–100% ethanol, stained tissues were observed with a microscope.
For the measurement of accumulation of hepatic lipid droplets, oil red O staining was performed with liver tissues. After adjusting with propylene glycol to avoid carrying water into oil red O solution, cut-liver tissues were stained with pre-warmed oil red O solution for 10 min. After staining with 85% propylene glycol for 5 min, stained tissues were observed with a microscope.
Statistical analysis
The data are presented as means ± standard error of the mean. Statistical analyses were conducted by employing Statistical Analysis Systems statistical software package (IBM SPSS Statistics Version 25.0; IBM Corporation, Armonk, NY, USA). A graph was created by using GraphPad Prism software 5.0 (GraphPad, San Diego, CA, USA). Significance was determined by one-way analysis of variance, least significance difference and Duncan post-hoc test for multiple comparisons. Differences were considered significant at P < 0.05.
RESULTS
Induction of ABCG1 and apoE by ellagic acid
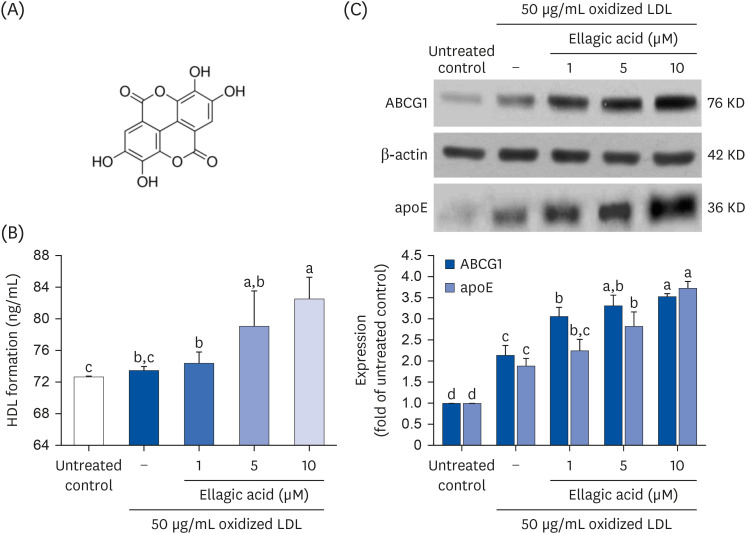
Cytotoxicity and lipid peroxidation did not take place in J774A1 macrophages incubated with ≤ 10 μM ellagic acid for 24 h (data not shown). Ellagic acid dose-dependently promoted the HDL formation from lipid-laden cells (Fig. 1B). In addition, ABCG1 was significantly induced in 50 μg/mL oxidized LDL-added macrophages for 18 h (Fig. 1C). Such induction was further up-regulated by the 18 h-treatment with ellagic acid. Our previous study showed that ≥ 1 μM ellagic acid promoted cholesterol efflux outside macrophages exposed to oxidized LDL [24]. This was most likely due to ellagic acid-triggered induction of ABC transporters. Furthermore, oxidized LDL enhanced macrophage production of apoE, which was further accelerated by adding ≥ 1 μM ellagic acid to oxidized LDL-exposed macrophages (Fig. 1C).
Fig. 1
Chemical structure of ellagic acid (A), HDL formation (B), and up-regulation of oxidized LDL-induced cellular ABCG1 expression and apoE secretion (C) by ellagic acid. J774A1 macrophages were treated with 1–10 μM ellagic acid for 18 h-stimulation with 50 μg/mL oxidized LDL. HDL formation was measured by using an enzyme-linked immunosorbent assay kit (B). For the measurement of ABCG1 expression and apoE secretion, total cell lysates and culture media were subjected to Western blot analysis with a primary antibody against ABCG1 or apoE (C). β-Actin was used as an internal control. The bar graphs represent quantitative results of blots obtained from a densitometer. Respective values (mean ± standard error of the mean, n = 3) in bar graphs not sharing a small letter are significantly different at P < 0.05.
HDL, high-density lipoprotein; LDL, low-density lipoprotein; ABCG1, ATP-binding cassette transporter G1; apoE, apolipoproteins E.
General characteristics of animals
Following the experimental design described in Fig. 2A, WT C57BL/6N mice and apoE KO mice were fed atherogenic Paigen diet for 10 weeks concurrently with oral administration of 10 mg/kg ellagic acid. The weight gain curve over 10 weeks and the end point weight gain after 10-weeks of dietary regimen were shown in Table 1, Fig. 2B. After the diet intervention, the final BW and average daily gain (ADG) significantly decreased in apoE KO mice, as compared to WT mice (Table 1). The oral administration of ellagic acid did not influence the weight gain pattern (Table 1, Fig. 2B). Since the average daily feed intake (ADFI) of apoE KO mice was not different from that of WT mice, the food efficiency ratio (FER = ADG/ADFI) declined in apoE KO mice (Table 1). The liver wet weight of apoE KO mice was less than that of WT mice, while the heart weight was larger (Table 1). The organ weights of apoE KO mice were not changed by oral supplementation of ellagic acid.
Fig. 2
Schematic illustration of animal experimental design/timeline (A) and BW gain (B). WT mice (C57BL/6N) and apoE KO mice were fed with atherogenic Paigen diet for 10 weeks with and without oral administration of 10 mg/kg ellagic acid daily. BW was measured weekly for 10 weeks. Graph was shown the average BW of each group as a time-response of weeks. Statistical differences were shown as asterisks (*). Values (n = 16, each diet group) on line graphs indicate a significant difference at *P < 0.05, compared to WT.
BW, body weight; WT, wild type; apoE KO, apolipoproteins E knockout.
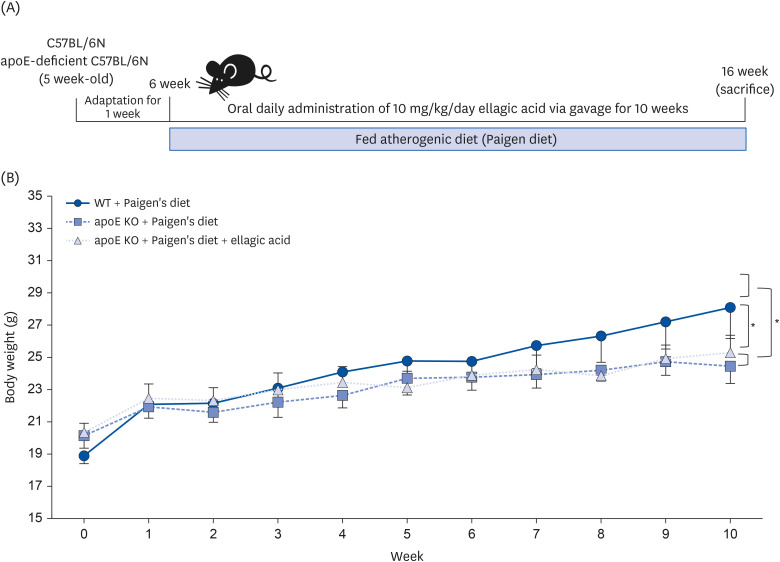
Table 1
Effect of ellagic acid on growth, food intake, and organ weights
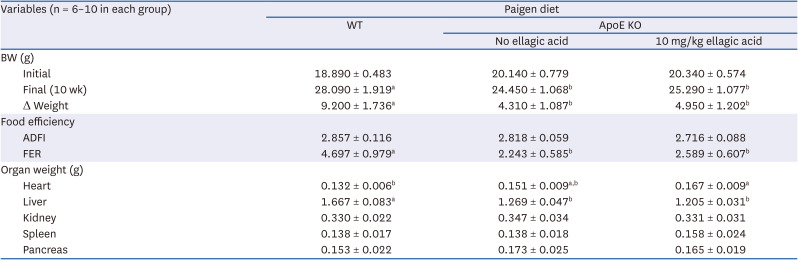
WT C57BL/6 mice (6 weeks of age, 20 g) and homozygous apoE KO mice were fed atherogenic Paigen diet (40 kcal% fat, 1.25% cholesterol, 0.5% cholic acid) for 10 weeks. One diet group was orally supplemented with 10 mg/kg ellagic acid. BW was measured weekly, and food intake was daily checked by measuring the amount of provided diets and remained diet after one day. After 10 weeks of the experimental period, each organ of heart, liver, kidney, spleen, and pancreas was dissected and measured by scale. All data were analyzed with a 1-way analysis of variance, followed by Duncan’s post-hoc tests. Respective values (n = 16, each diet group) in the same row not sharing the same superscript differ, P < 0.05.
WT, wild type; ApoE KO, apolipoproteins E knockout; BW, body weight; ADFI, average daily feed intake; FER, feed efficiency ratio.
Lipid-lowering effect of ellagic acid
This study investigated plasma lipid profiles of Paigen diet-fed apoE KO mice supplemented with ellagic acid (Table 2). Plasma levels of TC and LDL-C were markedly elevated in apoE KO mice fed Paigen diet, as compared to WT mice fed Paigen diet (Table 2). Plasma levels of TG and VLDL were not significantly different from those of WT mice. In contrast, plasma HDL-C level decreased in apoE-deficient mice. When 10 mg/kg ellagic acid was administrated to apoE KO mice, plasma levels of these lipids were reversed. As a result, the AI declined in ellagic acid-treated apoE KO mice (Table 2). It should be noted that ellagic acid increased plasma levels of TG and VLDL.
Table 2
Changes of plasma lipid levels and AI by ellagic acid supplementation
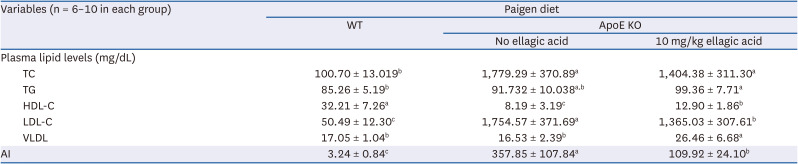
WT C57BL/6 mice (6 weeks of age, 20 g) and homozygous apoE KO mice were fed atherogenic Paigen diet (40 kcal% fat, 1.25% cholesterol, 0.5% cholic acid) for 10 weeks. One diet group was orally supplemented with 10 mg/kg ellagic acid. Plasma levels of TC, TG, and HDL-C, and the levels of LDL-C and VLDL were calculated by formulas. Equation: LDL= TC – (HDL – TG/5), VLDL = TG/5 and AI = (TC – HDL-C)/HDL-C. All data were analyzed with a one-way analysis of variance, followed by Duncan’s post-hoc tests. Respective values (n = 16, each diet group) in the same row not sharing the same superscript differ, P < 0.05.
WT, wild type; ApoE KO, apolipoproteins E knockout; TC, total cholesterol; TG, triglycerides; HDL-C, high-density lipoprotein-cholesterol; LDL-C, low-density lipoprotein-cholesterol; VLDL, very low-density lipoprotein; AI, atherosclerosis index.
Elevation of peripheral cholesterol efflux by ellagic acid
The cellular membrane transporter ABCA1 and ABCG1 facilitate active efflux of cholesterol [26]. This study examined whether ellagic acid favorably manipulated the transcription of peripheral ABCA1 and ABCG1 in apoE KO mice. The proteins of ABCA1 and ABCG1 were induced in peritoneal macrophages isolated from apoE KO mice fed Paigen diet (Fig. 3A). When 10 mg/kg ellagic acid was orally administrated to apoE KO mice, the induction of these transporters was further promoted. In addition, the SR-B1 expression was elevated in peritoneal macrophages of apoE KO mice, which was also further increased by supplying ellagic acid to mice (Fig. 3B).
Fig. 3
Effects of ellagic acid on transporter proteins responsible for peripheral cholesterol efflux. WT mice and apoE KO mice were fed atherogenic Paigen diet for 10 weeks with oral administration of 10 mg/kg ellagic acid daily. After the experimental period for 10 weeks, peritoneal macrophages were isolated from each mouse. Whole cell lysates were subject to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot with a specific antibody against ABCA1, ABCG1, and SR-B1 (A, B). β-Actin was used as an internal control. Respective values (mean ± standard error of the mean, n = 3) in bar graphs not sharing a small letter are significantly different at P < 0.05. Transcriptional levels of ABCA1, ABCG1, and SR-B1 were determined by using semi-quantitative reverse transcription polymerase chain reaction assay (C). GAPDH was used as a housekeeping gene for the co-amplification with ABCA1, ABCG1, and SR-B1.
WT, wild type; apoE KO, apolipoproteins E knockout; ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; SR-B1, scavenger receptor-B1; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
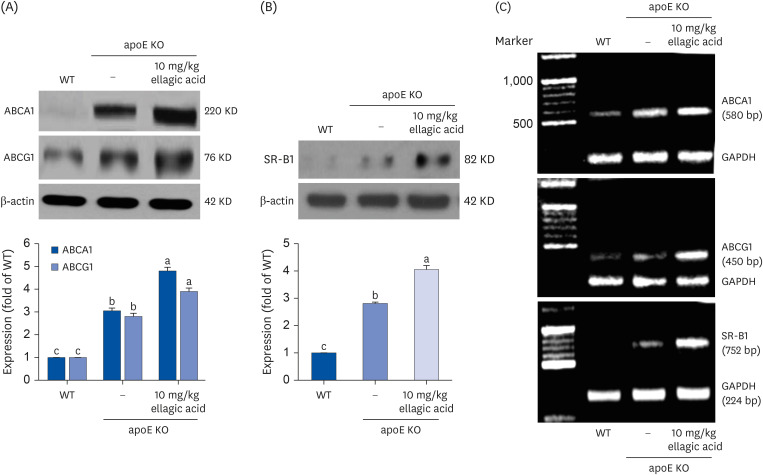
This study further examined the transcriptional levels of the cholesterol transporters of ABCA1, ABCG1 and SR-B1 in peritoneal macrophages isolated from high cholesterol diet-fed apoE KO mice, evidenced by semi-quantitative RT-PCR analysis. The transcription of these transporters was upregulated in Paigen diet-fed apoE KO mice (Fig. 3C). When the apoE KO mice received 10 mg/kg ellagic acid, the transcriptional levels of these proteins were highly elevated. Accordingly, ellagic acid may promote macrophage cholesterol efflux through inducing these cholesterol transporters at the transcriptional levels.
Blockade of induction of lipid transfer proteins by ellagic acid
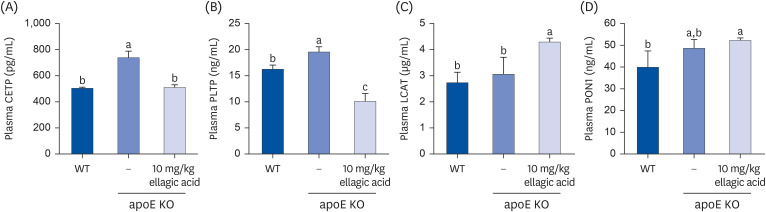
This study examined whether ellagic acid modulated plasma lipid levels through influencing the RCT process. Plasma levels of CETP and PLTP involved in RCT process were elevated in Paigen diet-fed mice lacking apoE gene, compared to those of WT mice (Fig. 4A and B). Such elevation was substantially reduced by supplementing 10 mg/kg EA for 10 weeks to apoE KO mice. On the contrary, oral administration of 10 mg/kg ellagic acid enhanced plasma LCAT level of apoE KO mice fed a high-cholesterol Paigen diet (Fig. 4C). Furthermore, there was a small increase in plasma PON1 level by apoE gene depletion (Fig. 4D). However, the supply of ellagic acid markedly enhanced the plasma PON1 level. Accordingly, ellagic acid may accelerate RCT process, leading to a reduction of plasma cholesterol level in apoE KO mice.
Fig. 4
Effects of ellagic acid on plasma levels of CETP, PLTP, LCAT, and PON1. WT mice and apoE KO mice were fed atherogenic Paigen diet for 10 weeks with and without oral administration of 10 mg/kg ellagic acid daily. Plasma levels of CETP, PLTP, LCAT, and PON1 were measured by using ELISA kits. All the ELISA procedures were followed according to the manufacturer’s instructions. Means (mean ± standard error of the mean, n = 3) in bar graphs without a common letter differ at P < 0.05.
CETP, cholesterol ester transfer protein; PLTP, phospholipid transport protein; LCAT, lecithin-cholesterol acyltransferase; PON1, paraoxonase-1; WT, wild type; apoE KO, apolipoproteins E knockout; ELISA, enzyme-linked immunosorbent assay.
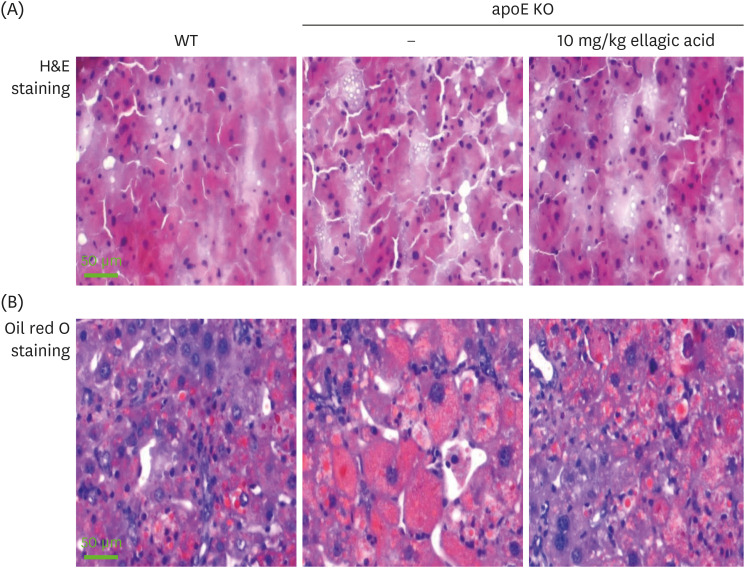
Inhibition of hepatic lipid accumulation by ellagic acid
This study investigated that ellagic acid attenuated hepatic lipid accumulation from circulation via RCT process. There was an increase in hepatic lipid droplets in apoE KO mice, evidenced by H&E staining (Fig. 5A). In addition, heavy oil red O staining was observed in apoE KO mice, which was diminished by supplementing ellagic acid (Fig. 5B).
Fig. 5
Effects of ellagic acid on hepatic morphology (A) and lipid accumulation (B). WT mice and apoE KO mice were fed atherogenic Paigen diet for 10 weeks with oral administration of 10 mg/kg ellagic acid daily. Liver tissues were dissected and cut by 6 μm thickness with microtomes. For the hepatic morphology, the tissues were stained with H&E (A). Hepatic lipid accumulation was confirmed by staining with oil red O (B). Liver tissues were observed by microscopy with 200× magnification (scale bar = 50 μm).
WT, wild type; apoE KO, apolipoproteins E knockout; H&E, hematoxylin and eosin.
Effects of ellagic acid on induction of hepatic receptors
This study examined the protein expression of the oxidized LDL receptor LOX-1 and the hepatic HDL receptor SR-B1 in hepatic tissues. The apoE gene deletion and high cholesterol feeding elevated the induction of hepatic LOX-1 and SR-B1 (Fig. 6A and B). Oral administration of ellagic acid to apoE KO mice minimally increased hepatic LOX-1 induction (Fig. 6A). Ellagic acid highly augmented the induction of LOX-1 and SR-B1 of apoE KO mice exposed to high cholesterol diet (Fig. 6B), indicating that ellagic acid may improve the removal of oxidized LDL and intake of HDL-C to the liver.
Fig. 6
Effects of ellagic acid on expression of hepatic lipoprotein receptors. WT mice and apoE KO mice were fed atherogenic Paigen diet for 10 weeks with and without oral administration of 10 mg/kg ellagic acid daily. After the experimental period for 10 weeks, proteins and total mRNA of liver tissues were isolated. Western blot analysis showing protein levels of lectin-like oxidized LOX-1 and SR-B1 (A, B). Liver tissue extracts were subject to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot with a specific antibody against LOX-1 and SR-B1. β-Actin was used as an internal control. The bar graphs (mean ± standard error of the mean, n = 3) represent quantitative results of blots obtained from a densitometer. Transcription of SR-B1 and LDLR was determined by using real-time reverse transcription polymerase chain reaction with each primer. GAPDH was used as a housekeeping gene for the co-amplification with SR-B1 and LDLR. Respective values in bar graphs not sharing a small letter are significantly different at P < 0.05.
WT, wild type; apoE KO, apolipoproteins E knockout; LOX-1, low-density lipoprotein receptor-1; SR-B1, scavenger receptor-B1; LDLR, low-density lipoprotein receptor; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
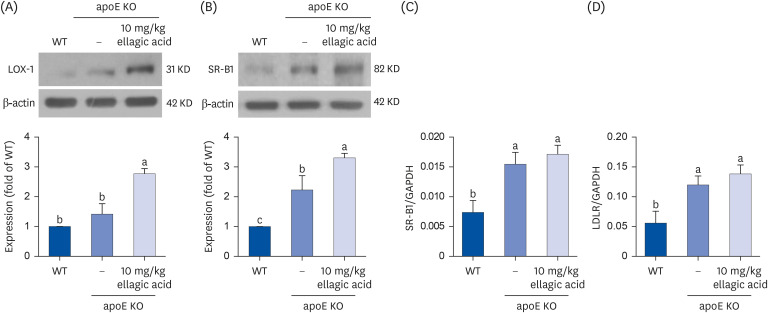
This study attempted to determine that ellagic acid activated hepatic SR-B1 and LDLR of apoE KO mice at transcriptional stages, evidenced by real-time PCR analysis. The hepatic transcription of SR-B1 and LDLR increased in Paigen diet-fed apoE KO mice, compared to that of WT mice (Fig. 6C and D). The hepatic transcription of these receptors of SR-B1 and LDLR continued to increase in apoE KO mice receiving 10 mg/kg ellagic acid (Fig. 6C and D). Thus, ellagic acid may lower plasma level of non-HDL cholesterol in hypercholesterolemic apoE KO mice.
Elevation of hepatic cholesterol uptake by ellagic acid
This study examined whether ellagic acid positively influenced the production of the HDL component apoA1 in apoE KO mice. The Paigen diet lowered plasma apoA1 level and hepatic apoA1 production in mice lacking apoE (Fig. 7A and B). In contrast, oral administration of ellagic acid enhanced the apoA1 secretion in the circulation and hepatic apoA1 expression in apoE KO mice (Fig. 7C). The hepatic production of apoA1 by ellagic acid was achieved at transcriptional level. In addition, the transcription of the AIBP protein secreting from liver was highly enhanced by supplying ellagic acid to apoE KO mice fed with Paigen diet (Fig. 7D). Accordingly, ellagic acid may stimulate hepatic apoA1 production and secretion in atherosclerosis-prone apoE KO mice.
Fig. 7
Effects of ellagic acid on production and binding of apoA1. WT mice and apoE KO mice were fed atherogenic Paigen diet for 10 weeks with and without oral administration of 10 mg/kg ellagic acid daily. After the experimental period for 10 weeks, blood was collected. Proteins and total mRNA of liver tissues were collected. Plasma and liver tissue extracts were subject to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot with a specific antibody against apoA1. β-Actin was used as an internal control. The bar graphs (mean ± standard error of the mean, n = 3) represent quantitative results of blots obtained from a densitometer. Transcription of apoA1 and AIBP was determined by using real-time reverse transcription polymerase chain reaction with each primer. GAPDH was used as a housekeeping gene for the co-amplification with apoA1 and AIBP. Respective values in bar graphs not sharing a small letter are significantly different at P < 0.05.
ApoA1, apolipoprotein A1; WT, wild type; apoE KO, apolipoproteins E knockout; AIBP, apoA1 binding protein; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
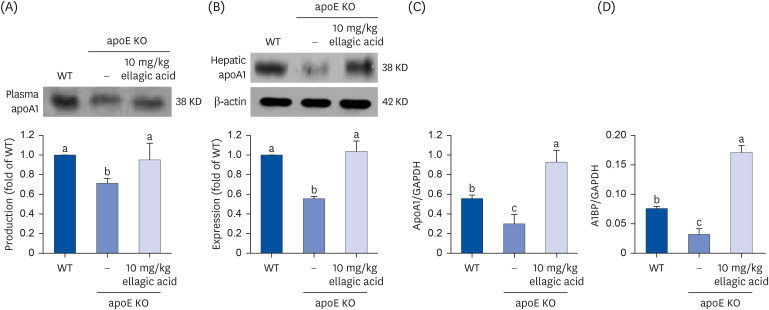
Effect of ellagic acid on hepatic cholesterol efflux
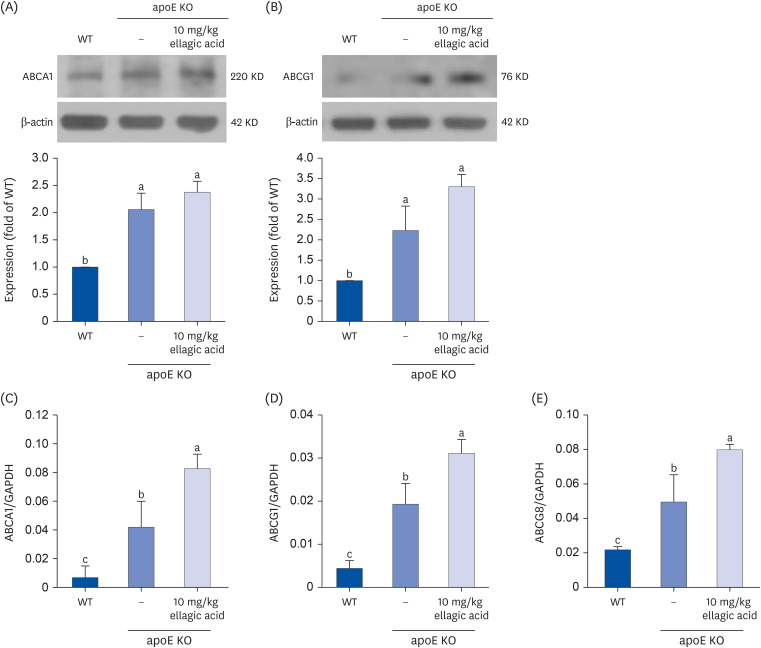
To determine whether ellagic acid promotes cholesterol efflux in hepatic tissues, this study examined the protein expression and transcription of ABCA1 and ABCG1. The hepatic ABCA1 and ABCG1 were induced in apoE KO mice fed Paigen diet (Fig. 8A and B). In apoE KO mice supplemented with 10 mg/kg ellagic acid, these proteins were highly boosted. Furthermore, the transcription of these transporters of ABCA1, ABCG1 and ABCG8 was enhanced in Paigen diet-fed apoE KO mouse liver (Fig. 8C-E). When 10 mg/kg ellagic acid was administrated to the apoE KO mice, the transcriptional levels of these proteins continued to increase.
Fig. 8
The effects of ellagic acid on hepatic induction of cholesterol transporters responsible for cholesterol efflux. WT mice and apoE KO mice were fed atherogenic Paigen diet for 10 weeks with and without oral administration of 10 mg/kg ellagic acid daily. After the experimental period for 10 weeks, proteins and total mRNA of liver tissues were isolated. Protein expressions of ABCA1 and ABCG1 were measured by Western blot analysis (A, B). Liver tissue extracts were subject to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and Western blot with a specific antibody against ABCA1 and ABCG1. β-Actin was used as an internal control. The bar graphs (mean ± standard error of the mean, n = 3) represent quantitative results of blots obtained from a densitometer. Transcription of ABCA1, ABCG1, and ABCG8 was determined by using real-time reverse transcription polymerase chain reaction with each primer (C-E). GAPDH was used as a housekeeping gene for the co-amplification with ABCA1, ABCG1, and ABCG8. Respective values in bar graphs not sharing a small letter are significantly different at P < 0.05.
WT, wild type; apoE KO, apolipoproteins E knockout; ABCA1, ATP-binding cassette transporter A1; ABCG1, ATP-binding cassette transporter G1; ABCG8, ATP-binding cassette transporter G8; GAPDH, glyceraldehyde 3-phosphate dehydrogenase.
DISCUSSION
The nine major findings were extracted from this study employing ellagic acid. 1) Ellagic acid further up-regulated induction of HDL, ABCG1 and apoE in oxidized LDL-exposed macrophages. 2) Oral administration of ellagic acid did not change the weight gain pattern, average daily food intake and FER of Paigen diet-fed apoE KO mice. 3) Ellagic acid favorably influenced plasma levels of atherogenic lipids in apoE KO mice. 4) Protein induction and transcription of ABCA1, AGCG1 and SR-B1 were elevated by ellagic acid in mouse peritoneal macrophages isolated from apoE KO mice. 5) Plasma levels of CETP and PLTP elevated in apoE KO mice was substantially reduced by supplementing ellagic acid, while plasma levels of LCAT and PON1 was elevated. 6) A marked increase in hepatic lipid droplets in Paigen diet-fed apoE KO mice was diminished in ellagic acid-treated mice. 7) Ellagic acid appeared to further increase the transcription of hepatic SR-B1 and LDLR elevated in Paigen diet-fed apoE KO mice. 8) The apoA1production and AIBP expression highly increased at transcriptional levels in Paigen diet-fed apoE KO mice receiving ellagic acid. 9) Ellagic acid boosted the induction and transcription of hepatic ABCA1and AGCG1 in apoE KO mice. Therefore, ellagic acid improved RCT functionality and HDL function in diet-induced atherogenesis of apoE KO mice.
ApoE acts as a ligand for LDLR family members to mediate the clearance of apoB-containing TG-rich lipoproteins [27]. In addition, apoE is involved in cholesterol efflux in peripheral tissues, especially from cholesterol-loaded macrophages and in RCT process [16]. ApoE present in plasma lipoproteins is responsible for the clearance of chylomicrons and VLDL remnants by the liver [18]. Thus, the apoE absence would impair clearance of cholesterol-enriched lipoproteins, resulting high levels of plasma cholesterol. Furthermore, the apoE absence renders animals more susceptible to diet-induced atherosclerosis [919]. A study reports that apoE-null mice fed the high-fat/cholesterol/cholate Paigen diet show severe hypercholesterolemia, and lipid deposits were detected in all the organs [28]. Consistently, this study revealed that apoE KO mice fed on Paigen diet accelerated the accumulation of cholesterol-rich lipoproteins, compared with WT mice. A limited number of studies have been performed in the apoE-depleted mice to explore the role of dietary components including fatty acids and protein sources on lipoprotein metabolism and atherosclerosis.
Numerous studies have demonstrated that several phenolic compounds act as potential agents against atherosclerosis in preclinical and clinical studies [293031]. Based on preclinical and clinical evidence, the main actions of phenolic compounds is known to be associated with inhibition of LDL oxidation, anti-inflammatory effects and reduction of platelet activity [2932]. Ellagic acid is phenolic phytochemicals that display potent antioxidant and free radical scavenging activities, thus preventing oxidative stress-related diseases [212233]. Ellagic acid exhibits vascular health benefits via diverse mechanisms for anti-atherogenic, anti-thrombotic, anti-inflammatory and anti-angiogenic effects [22]. Our previous study has shown that ellagic acid blocks foam cell formation and boosts cholesterol efflux in lipid-laden foam cell macrophages [24]. The present study examined whether ellagic acid enhanced the HDL function through improving the RCT system. This study examined the combined effects of apoE gene deletion and high cholesterol on the RCT pathway and hepatic lipid clearance. Ellagic acid alleviated hypercholesterolemia, and instead enhanced plasma HDL-C level with PON1 in apoE KO mice fed a high-cholesterol diet. Subsequently, ellagic acid improved RCT functionality and HDL function in diet-induced atherogenesis of apoE-depleted mice.
In the RCT systems, HDL metabolism begins with binding of cholesterol effluxed from peripheral tissues or cells via the transporters of ABCA1 or ABCG1 [891134]. Ellagic acid markedly increased the protein expression of ABCA1 and ABCG1 at the transcriptional levels in peritoneal macrophages isolated from Paigen diet-fed apoE KO mice. Like oxidized LDL, the cholesterol-rich Paigen diet elevated the expression of ABC transporters of apoE KO mouse macrophages. In fact, ellagic acid induced apoE protein and HDL formation in oxidized LDL-exposed macrophages. Collectively, ellagic acid may be a substitute for apoE, which could be potential therapeutic replacement for preventing atherosclerosis. Macrophage apoA1 can protects against atherosclerosis by upregulating ABC transporters, stimulating cholesterol efflux and RCT [20]. Although this study did not examine the apoA1 level in peritoneal macrophages of apoE KO mice, the expression of ABC A1 and ABCG1 was highly enhanced. On the other hand, ellagic acid led to decreased CETP activity and PLTP activity and increased LCAT activity in Paigen diet-fed apoE KO mice, which could be recognized as protective factors. These findings indicate that ellagic acid was effective in forming pre-β-HDL and larger HDL particles through efflux of cellular lipids to increased lipid-free apoA1.
This study found that ellagic acid diminished an increase in hepatic lipid droplets in apoE KO mice fed Paigen diet. SR-B1 facilitates cholesterol efflux from peripheral tissues back to liver, functioning as a HDL receptor [35]. Also, hepatic SR-BI plays an essential role in controlling the utilization of HDL cholesterol for biliary secretion [36]. Ellagic acid stimulated the SR-B1 induction in apoE KO mouse macrophages exposed to high-cholesterol diet, possibly enhancing cholesterol delivery to liver for utilization of hepatic cholesterol. In addition, ellagic acid elevated hepatic SR-B1, leading to increased cholesterol clearance via HDL-relevant influx of cholesteryl esters. Interestingly, ellagic acid induced LDLR for the LDL-C clearance, which may reduce plasma cholesterol in apoE KO mice. The LOX-1 induction of ellagic acid may alleviate the progression of atherosclerosis via clearance of oxidized LDL from circulation [37]. On the other hand, ellagic acid increased the hepatic expression of ABC transporters and apoA1 in apoE KO mice fed Paigen diet. ABCA1 plays a key role in apoA1-dependent cholesterol efflux and cholesterol homeostasis in liver as a major source of plasma HDL-C [38]. Accordingly, ellagic acid may compensate for the absence of apoE in maintaining cholesterol homeostasis in the liver and circulation.
In summary, the current report demonstrated that ellagic acid improved RCT functionality and HDL function in diet-induced atherogenesis of cholesterol diet-fed apoE KO mice. Ellagic acid elevated induction of ABC transporters and apoA1 in apoE KO mice, possibly forming HDL particles via efflux of cellular lipids. Subsequently, ellagic acid elevated plasma HDL-C reduced in Paigen diet-fed apoE KO mice and conversely reduced LDL-C. Ellagic acid diminished plasma levels of lipid transfer proteins for delivery of cholesteryl esters and phospholipids. Ellagic acid stimulated the SR-B1 induction in peritoneal macrophages of apoE KO mice, enhancing cholesterol delivery to the liver for utilization of hepatic cholesterol. While there is good evidence on ellagic acid supporting the anti-atherogenic effects in animal models, the evidence on humans is much scarcer.
 XML Download
XML Download