

 PDF
PDF Citation
Citation Print
Print



Introduction
Inflammatory myofibroblastic tumor (IMT) is a rare and independent soft-tissue tumor that originates from myofibroblastic cells with infiltration of inflammatory cells (mainly lymphocytes and plasma cells). IMT was previously thought to be reactive inflammatory hyperplasia and is now classified as a mesenchymal neoplasm with intermediate biologic potential [1,2]. The abdominopelvic region is the most common location of IMT, although it can occur at any site. IMT mainly affects children and adolescents, with an average age of approximately 10 years; however, it can occur at any age [3,4]. This condition is slightly more common in women [5]. A highly invasive type of IMT, epithelioid inflammatory myofibroblastic sarcoma (EIMS), has recently been reported. Most EIMS occurred in young men and almost only occurred in the omentum and mesentery in the abdominopelvic region. EIMS is associated with more aggressive behaviors [6,7].
Recent studies have confirmed that anaplastic lymphoma kinase (ALK) gene rearrangement on chromosome 2P23 is present in approximately 50% of patients with IMT, resulting in ALK expression. In the remaining IMT patients, fusions involving ROS1, NTRK, and RET were also observed, suggesting that tyrosine kinase rearrangements play an important role in most IMT tumors [8–12].
Complete surgical resection is the primary treatment option for patients with IMT. However, local recurrence may occur after the initial surgery, with a low risk of distant metastases. No standard systemic treatment is available for patients with unresectable or metastatic IMT. Although steroids and chemotherapy (anthracycline/ifosfamide, methotrexate, and vinorelbine/vinblastine) have shown inconsistent efficacy, recent studies have shown that ALK inhibitors are highly effective in patients with ALK-positive advanced IMTs [13].
Most published trials and data have arisen from pediatric studies [14–17]. However, adult IMT is rare and has rarely been reported. The clinical and pathological features of adult IMT are unclear, as are the systemic treatment options and responses to ALK inhibitors. Therefore, this retrospective study aimed to explore the clinicopathological characteristics and treatment of adult IMT patients.
Materials and Methods
1. Patients selection
The inclusion criteria were as follows: aged ≥ 18 years old, histologically confirmed diagnosis of IMT by the Department of Pathology, Fudan University Shanghai Cancer Center (FUSCC), involving any anatomical compartment, and had received treatment in FUSCC between 2006 and 2021. The exclusion criteria were as follows: insufficient follow-up data, including unknown clinicopathological characteristics, no treatment record, or no follow-up information. Clinical and pathological data, treatments, and outcomes of the enrolled patients were collected and analyzed.
2. Treatment scheme for IMTs in FUSCC
The treatment strategy of IMT in FUSCC was in accordance with the National Comprehensive Cancer Network (NCCN) Clinical Practice guideline for soft tissue sarcoma. At diagnosis, the tumor extent was assessed with computerized tomography and/or magnetic resonance imaging. For localized disease, complete surgical resection was the primary treatment. No adjuvant therapy was given after radical surgery. For metastatic or inoperable IMTs, systemic therapy would be given. Due to the lack of a standard of care, the systemic therapy regimen was given per physician’s choice. ALK immunohistochemical analysis (IHC) and ALK fluorescence in situ hybridization (FISH) test were routinely performed. Next-generation sequencing (NGS) was performed on paraffin-embedded and/or fresh frozen tissue depending on tumor tissue availability and patients’ economical capability, as the costs of NGS are very high and are not covered by national medical insurance. Generally, for patients with ALK or other gene translocation, targeted therapy such as ALK inhibitor was given as the first-line treatment. For patients resistant to first-generation tyrosine kinase inhibitors second-generation drugs were applied according to the drug availability and patients’ affordability. For patients without ALK or other gene translocation, chemotherapy was given as the first-line treatment, mainly anthracycline-based regimens and methotrexate±vinorelbine/vinblastine regimens.
Overall survival (OS) was calculated as the time from IMT diagnosis to death from any cause. Progression-free survival (PFS) was defined as the time from the initiation of systemic treatment to tumor progression or death. OS from the initiation of systemic treatment to death from any cause was described as OS for systemic treatment. Survival curves were calculated using the Kaplan-Meier method and compared using the log-rank test for univariate analysis. For efficacy evaluation, Response Evaluation Criteria in Solid Tumors (ver. 1.1) were used, which classified responses into complete response (CR), partial response (PR), stable disease (SD), and progressive disease (PD). The objective response rate (ORR) was calculated as CR+PR, and the disease control rate (DCR) was calculated as CR+PR+SD. Adverse reactions were assessed according to the National Cancer Institute–Common Terminology Criteria for Adverse Events (NCI-CTCAE4.0). SPSS ver. 22 software (IBM Corp., Armonk, NY) was used for statistical analysis of the relevant data.
Results
1. Patients’ characteristics
1) Clinical characteristics
Overall, 102 IMT patients were identified in our institution from 2006 to 2021. Among them, 39 patients were aged ≥ 18 years old. Nine patients with insufficient follow-up data were excluded. Therefore 30 patients were enrolled in the present study. The baseline clinical characteristics of the patients are presented in Table 1. The age range of the 30 adult patients was 21–77 years, with a median age of 38 years. Twelve patients were men (40.0%), and 18 were women (60.0%). The most common primary lesions were those in the abdominopelvic region (n=16; 53.3%), followed by the lungs (n=6, 20.0%), and retroperitoneum (n=2, 6.7%). There was one case with stomach, bladder, breast, liver, uterus, and rectum each. The most common presenting symptoms were abdominopelvic discomfort (18/30, 60%), fever (6/30, 20%), cough (5/30, 16.7%), and weight loss (4/30, 13.3%). Six patients were tested for carbohydrate antigen 125 (CA125) levels, and a moderate increase in CA125 levels was observed in four patients. Among the 30 adult IMT patients, seven had abdominal EIMS. There were three (42.9%) men and four (57.1%) women with a median age of 26 years (21–74 years).
Table 1
Baseline characteristics of 30 adult patients with inflammatory myofibroblastic tumor
![]()
Of the patients, 36.7% (11/30) had localized disease, and 63.3% (19/30) had metastatic or inoperable diseases. Among 19 patients with metastatic or inoperable diseases, primary tumors were located in the abdominopelvic region in 16 patients (one patient had liver, lung, bone, and lymph node metastasis; one patient had liver, pelvic, and lymph node metastasis; 14 patients had abdominal pelvic metastasis), lung in two patients (one patient had multiple lung metastases; one patient had mediastinal lymph node and abdominal pelvic metastasis), and in the rectum in one patient (liver metastasis). A total of 13.3% (4/30) of the patients had distant metastatic disease, with the liver (10%, 3/30) being the most common site of distant metastases.
2) Molecular characteristics
Twenty-seven patients were tested for ALK expression by IHC or for gene rearrangement by FISH. The ALK positivity rate was 81.5% (22/27). The consistency rate of ALK immunohistochemistry and gene rearrangement was 100%. Whereas the conventional IMTs showed diffuse cytoplasmic expression of ALK, the highly aggressive EIMSs exhibited a distinct pattern of nuclear membrane staining. All patients with EIMS were ALK-positive.
In patients with ALK-negative and ALK-positive tumors, the median age was 50 years (range, 34 to 77 years) and 30 years (range, 21 to 74 years), respectively. NGS was performed in four patients, and TPM3-ALK rearrangement, RANBP2 (RAN binding protein 2)–ALK rearrangement, and IGFBP5 (insulin like growth factor binding protein 5)–ALK rearrangement were detected; one patient had no targetable genetic abnormality. None of our patients were tested for programmed death-ligand 1 expression.
2. Patients’ treatment strategy
1) Surgery
Among 30 patients, 19 underwent radical surgery, while 11 did not, due to extensive disease. All seven patients with EIMS could not undergo radical surgical resection because of extensive lesions. Of the 19 patients who underwent radical surgery, eight (42.1%) had recurrence and/or metastasis after surgery. The time from surgery to relapse and/or metastasis of these eight patients ranged from 4.5–26.1 months (median time, 8.3 months; 95% confidence interval [CI], 7.3 to 9.4 months). These eight patients included two with lung IMT who had local recurrence or metastasis after surgery, four whose primary tumors were located in the abdominopelvic region, one patient whose primary tumor was located in the retroperitoneum who had abdominal and pelvic recurrence after surgery, and one patient with rectal IMT who had liver metastasis after surgery. The median disease-free survival has not been reached yet.
2) Systemic treatments and outcomes
In total, 16 patients with advanced ALK-positive IMT were treated with crizotinib (an oral small-molecule inhibitor of ALK, MET, and ROS1) at a dose of 250 mg twice daily. Crizotinib was administered as first-line therapy in 15 patients and as second-line therapy in one. The clinical characteristics and outcomes of these 16 patients are shown in Table 2.
Table 2
Characteristics and outcomes of 30 adult patients with inflammatory myofibroblastic tumor
| Patient No. | Sex/Age (yr) | Primary tumor site | Metastatic sites | Radical surgery | ALK IHC/ALK FISH/NGS tests | ALK inhibitor (BOR, PFS) | Other systemic therapy (BOR) | Outcome |
|---|---|---|---|---|---|---|---|---|
| 1 | M/34 | Abdominopelvic mesentery | Abdominopelvic mesentery | No | −/−/No actionable alterations | No | PLD+IFO (SD) | Lost |
| 2 | F/41 | Abdominopelvic mesentery | No | Yes | −/−/NA | No | No | No relapse, alive |
| 3 | M/56 | Retroperitoneum | Abdominopelvic mesentery | Yes | + (cytoplasmic)/+/NA | No | No | Relapse, dead |
| 4 | M/50 | Lung | No | Yes | −/−/NA | No | No | No relapse, alive |
| 5 | M/51 | Lung | No | Yes | NA | No | No | No relapse, alive, had nasopharyngeal carcinoma 8 years before the diagnosis of lung IMT |
| 6 | M/58 | Abdominopelvic mesentery | Abdominopelvic mesentery | Yes | −/−/NA | No | No | Relapse, lost |
| 7 | F/51 | Lung | No | Yes | NA | No | No | No relapse, developed pancreatic adenocarcinoma (PAC) 4.3 years after the diagnosis of lung IMT and died of PAC |
| 8 | F/32 | Bladder | No | Yes | + (cytoplasmic)/+/NA | No | No | Lost |
| 9 | M/26 | Stomach | No | Yes | + (cytoplasmic)/+/NA | No | No | No relapse, alive |
| 10 | M/25 | Lung | No | Yes | + (cytoplasmic)/+/TPM3-ALK translocation | No | No | No relapse, alive |
| 11 | F/52 | Retroperitoneum | No | Yes | + (cytoplasmic)/+/NA | No | No | No relapse, alive |
| 12 | F/41 | Breast | No | Yes | NA | No | No | No relapse, alive, developed thyroid cancer and cervical cancer 4.1 years and 7.7 years after the diagnosis of breast IMT, respectively |
| 13 | M/77 | Liver | No | Yes | −/−/NA | No | No | No relapse, alive |
| 14 | F/36 | Abdominopelvic mesentery | No | Yes | + (cytoplasmic)/+/NA | No | No | No relapse, alive |
| 15 | F/22 | Abdominopelvic mesentery | Abdominopelvic mesentery | No | + (cytoplasmic)/+/NA | Crizotiniba) (PR, > 13.3 mo) | No | Alive |
| 16 | F/47 | Abdominopelvic mesentery | Abdominopelvic mesentery, liver, lymph nodes | No | + (cytoplasmic)/+/NA | Crizotinibb) (CR, > 98.2 mo) | ADM (PD) | Alive, developed stage I lung adenocarcinoma 7.3 years after the diagnosis of pelvic IMT |
| 17 | M/22 | Abdominopelvic mesentery | Abdominopelvic mesentery | No | + (nuclear)/+/NA | Crizotiniba) (CR, > 53.1 mo) | No | Lost |
| 18 | F/24 | Lung | Lung, pleura | Yes | + (cytoplasmic)/+/NA | Crizotiniba) (PR, 18.3 mo) | Paclitaxel+ carboplatin (PD) | Dead |
| 19 | F/74 | Abdominopelvic mesentery | Abdominopelvic mesentery | No | + (nuclear)/+/NA | Crizotiniba) (PR, 20.8 mo), ceritinib (PR) | No | Lost |
| 20 | M/28 | Abdominopelvic mesentery | Abdominopelvic mesentery | No | + (nuclear)/+/NA | Crizotiniba) (PR, 9.0 mo), ceritinib (PR), lorlatinib (PR) | No | Dead |
| 21 | F/25 | Abdominopelvic mesentery and lymph nodes | Liver, lung, bone, mediastinal lymph nodes | No | + (nuclear)/+/NA | Crizotiniba) (PR, > 6.3 mo) | No | Lost |
| 22 | M/39 | Abdominopelvic mesentery | Abdominopelvic mesentery | No | + (nuclear)/+/NA | Crizotiniba) (PD, 1.3 mo) | No | Dead |
| 23 | F/51 | Abdominopelvic mesentery | Abdominopelvic mesentery and lymph nodes | No | + (nuclear)/+/NA | Crizotiniba) (SD, > 23.0 mo) | No | Alive |
| 24 | F/28 | Abdominopelvic mesentery | Abdominopelvic mesentery | Yes | + (cytoplasmic)/+/NA | Crizotiniba) (PD, 2.0 mo) | ADM+IFO (PD), anlotinib (SD) | Alive |
| 25 | F/21 | Abdominopelvic mesentery | Abdominopelvic mesentery and lymph nodes | Yes | + (cytoplasmic)/+/NA | Crizotiniba) (PR, > 30.2 mo) | No | Alive |
| 26 | F/56 | Abdominopelvic mesentery | Abdominopelvic mesentery | No | + (nuclear)/+/RANBP2-ALK translocation | Crizotiniba) (CR, > 19.4 mo) | No | Alive |
| 27 | F/28 | Uterus | Abdominopelvic mesentery and lymph nodes | No | + (cytoplasmic)/+/IGFBP5-ALK translocation | Crizotiniba) (PR, > 8.7 mo) | No | Alive |
| 28 | F/49 | Abdominopelvic mesentery | Abdominopelvic mesentery | Yes | + (cytoplasmic)/+/NA | Crizotiniba) (PR, > 6.1 mo) | No | Alive |
| 29 | F/51 | Lung | Abdominopelvic mesentery, mediastinal lymph nodes | Yes | + (cytoplasmic)/+/NA | Crizotiniba) (PR, > 2.3 mo) | No | Alive |
| 30 | M/28 | Rectum | Liver | Yes | + (cytoplasmic)/+/NA | Crizotiniba) (PR, 7.3 mo) | Apatinib (SD) | Alive |
ADM, doxorubicin; ALK, anaplastic lymphoma kinase; BOR, best of response, CR, complete response; F, female; FISH, fluorescence in situ hybridization; IFO, ifosfamide; IGFBP5, insulin like growth factor binding protein 5; IHC, immunohistochemical analysis; IMT, inflammatory myofibroblastic tumor; M, male; NA, not available; NGS, next-generation sequencing; PD, progressive disease; PFS, progression-free survival; PLD, pegylated-liposome doxorubicin; PR, partial response; RANBP2, RAN binding protein 2; SD, stable disease.
![]()
In 16 patients treated with crizotinib, 10 (62.5%) had PR, three (18.8%) achieved CR, one (6.3%) had SD, and two (12.5%) had PD. ORR was 81.3% (13/16), and DCR was 87.5% (14/16). Fig. 1 shows images of a patient with uterine IMT harboring an IGFBP5-ALK rearrangement who achieved PR after crizotinib treatment. The patient had an abdominopelvic region and lymph node metastases when first diagnosed. After treatment with crizotinib for > 10 months, the tumor showed PR. This patient was still receiving crizotinib treatment at the time of the analysis.
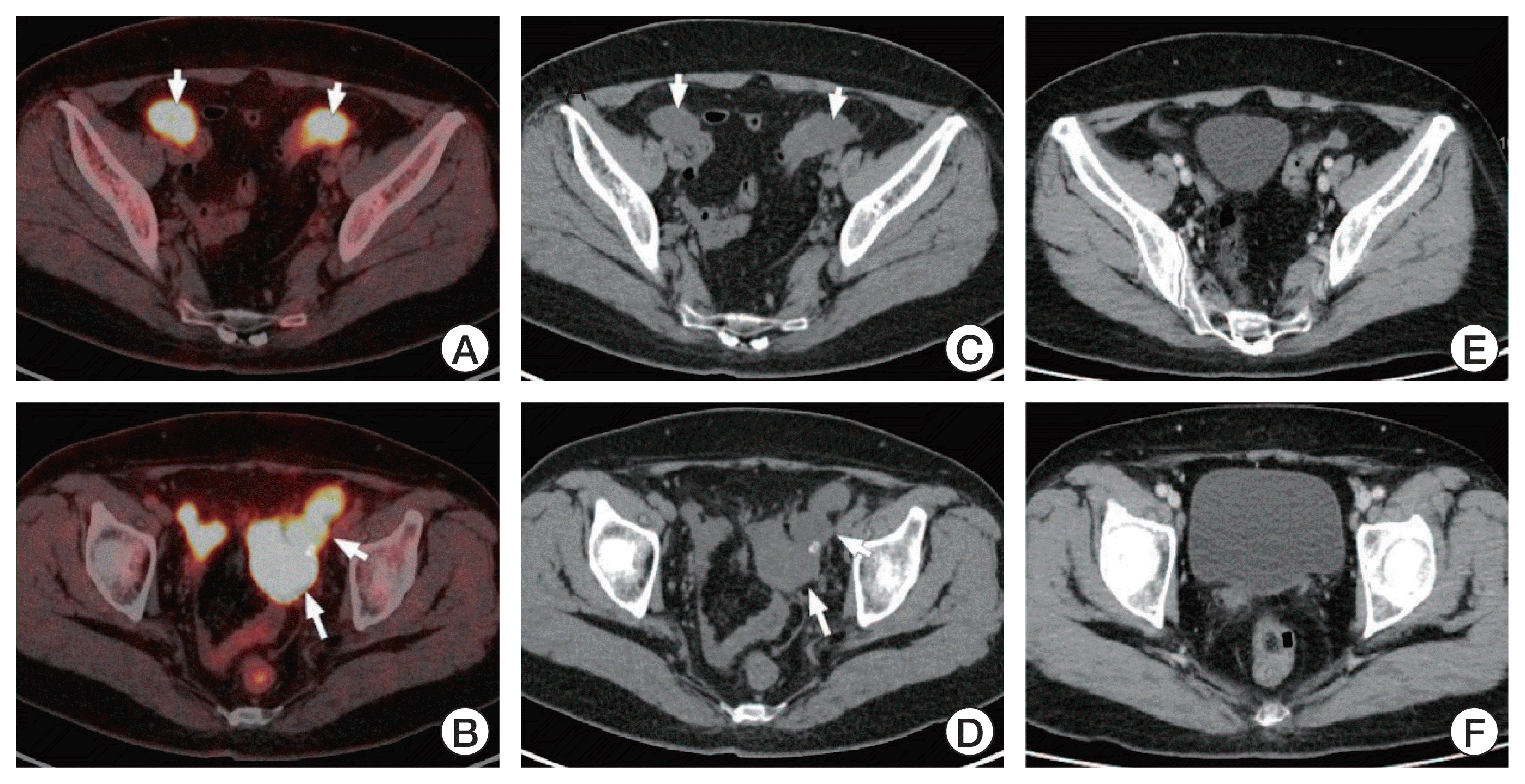
Fig. 1
The images of a patient with metastatic uterine inflammatory myofibroblastic tumor harboring IGFBP5-ALK rearrangement who achieved partial response after crizotinib therapy. (A, B) Positron emission tomography/computed tomographic images of a pelvic tumor at baseline. (C, D) Computed tomography images of a pelvic tumor at baseline. (E, F) A pelvic tumor that regressed after 9 months of crizotinib therapy. ALK, anaplastic lymphoma kinase; IGFBP5, insulin like growth factor binding protein 5.
![]()
In February 2022, the median follow-up time (from the time of crizotinib) was 23.0 months (95% CI, 16.5 to 29.5 months). Six patients progressed, eight patients were still on crizotinib treatment, and two were lost to follow-up (Fig. 2). The median PFS was 20.8 months (95% CI, unreached). The 1-year PFS rate was 70.7% (58.1%–83.3%).
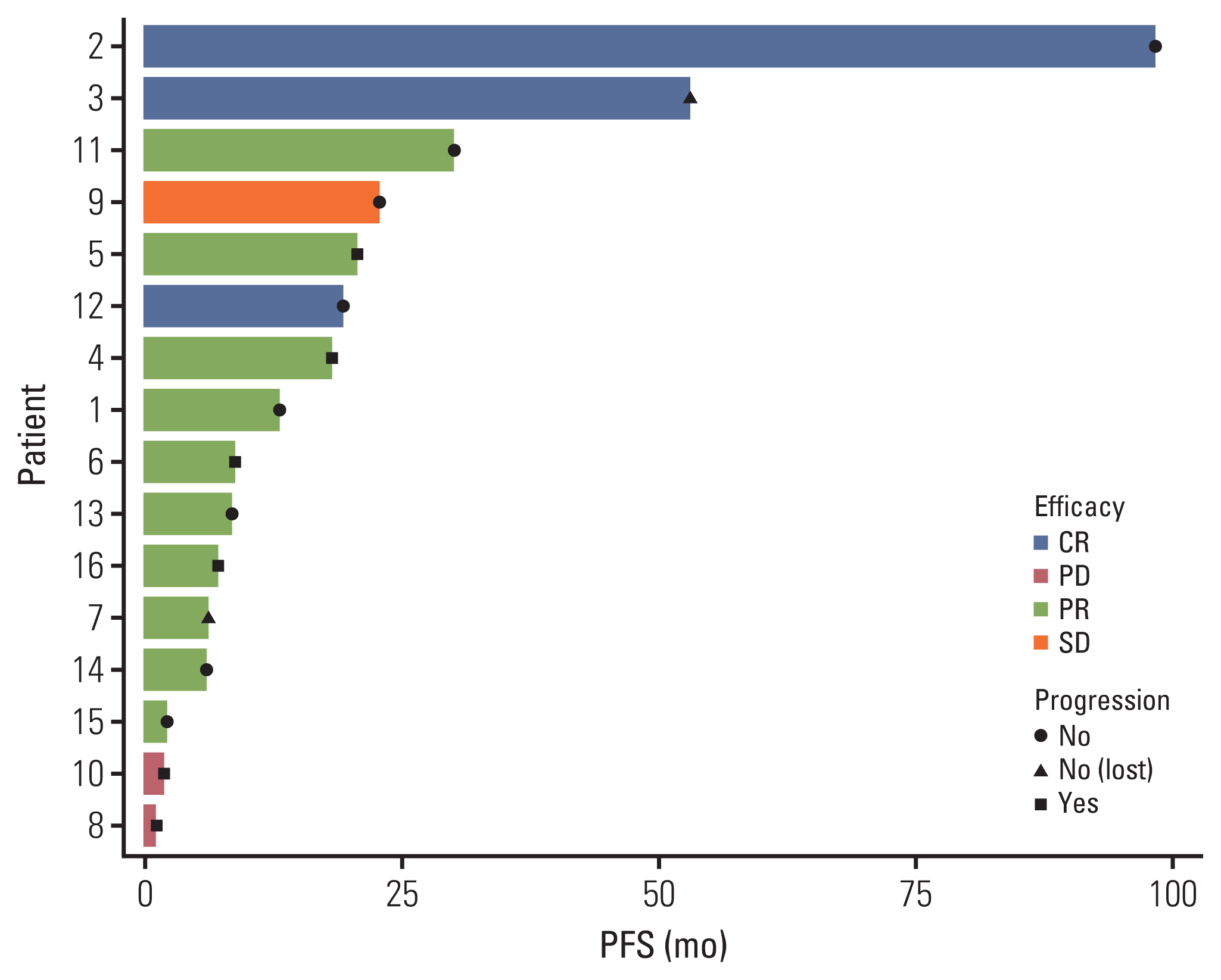
Of the six patients who progressed after crizotinib treatment, two were treated with ceritinib, a second-generation ALK inhibitor, with PR as their best response (patient Nos. 19 and 20). Patient No. 20 had achieved a PFS of 21.9 months for ceritinib, and a PFS of 6.2 months for subsequent lorlatinib. One patient (No. 18) was treated with paclitaxel and carboplatin (PD); one (No. 24) was treated with doxorubicin and ifosfamide (PD) and anlotinib (SD); one patient (No. 30) was treated with apatinib (SD), and one (patient No. 22) did not receive subsequent treatment.
By the time of the analyses, ten patients were still alive, three died, and three were lost to follow-up. The median OS for systemic treatment was not determined. The 2-year OS for systemic treatment rate was 78.1% (63.0%–93.2%). Among the seven patients with EIMS who were treated with crizotinib, three (43%) had PR, two had CR (29%), one (14%) had SD, and one (14%) had PD. ORR was 72%. The median PFS was 20.8 months (95% CI, unreached), with no significant difference compared to non-EIMS patients (p=0.987). Three patients progressed, and two died. One patient progressed rapidly and died 1.3 months after crizotinib administration.
The adverse reactions to crizotinib treatment were tolerable, and no grade III/IV adverse reactions occurred. The most common adverse reactions were grade I/II fatigue, nausea, diarrhea, and rash. One patient developed grade II interstitial pneumonia after medication, which resolved soon after drug withdrawal. Later, the dose of crizotinib was reduced to 200 mg twice daily for this patient, and no interstitial pneumonia occurred again.
3. Prognosis
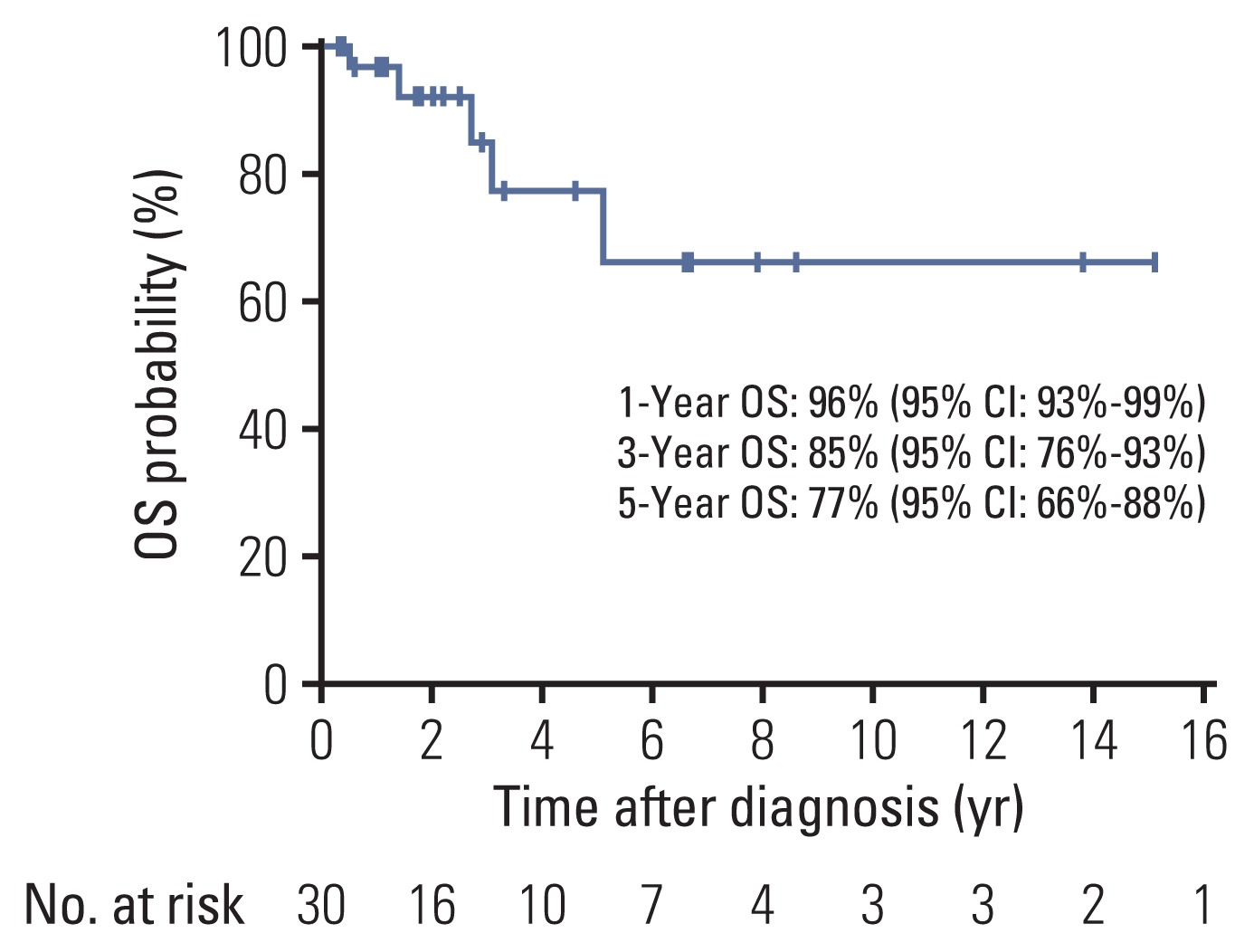
Up to February 2022, the median follow-up period (from the time of diagnosis) of all 30 patients was 30.0 months (95% CI, 13.6 to 46.4), and the 1-year, 3-year, and 5-year OS rates were 96% (95% CI, 93 to 99), 85% (95% CI, 76 to 93), and 77% (95% CI, 66 to 88), respectively (Fig. 3). Sex, age (< 40 vs. ≥ 40 years), primary tumor location, ALK status, and EIMS were not statistically correlated with OS in the univariate analyses. In all the 30 patients with ALK-positive and ALK-negative disease, the 1-year, 3-year, and 5-year OS rates were 100% and 100%, 100% and 95.2%, 77.8%, and 64.8%, respectively (p=0.254).
Four patients were diagnosed with multiple primary cancers (MPC). One patient (No. 16) had advanced ALK-positive disease and received crizotinib treatment for more than 8 years and developed stage I lung adenocarcinoma 7.3 years after the diagnosis of pelvic IMT. This patient’s abdominopelvic tumor showed complete remission during treatment with crizotinib, with enlargement of the pulmonary nodule, which was pathologically confirmed as lung adenocarcinoma with epidermal growth factor receptor (EGFR) exon 19 mutation. The lung lesions were treated using stereotactic radiotherapy. One patient (No. 5) had nasopharyngeal carcinoma 8 years before the diagnosis of lung IMT. One patient (No. 7) developed pancreatic adenocarcinoma (PAC) 4.3 years after the diagnosis of lung IMT and died of PAC. One patient (No. 12) developed thyroid cancer and cervical cancer 4.1 years and 7.7 years after the diagnosis of breast IMT, respectively.
Discussion
To the best of our knowledge, our study is the largest study on adult IMT, with 30 adult IMT patients included. The female-to-male ratio was 1.5 in our study, which was close to that in another study (1.4) [5]. IMT cases arising in the abdomen, retroperitoneum, or pelvis had been reported to be approximately 60%–65% [3]. It seems that adult IMT has a similar sex ratio and involved areas as pediatric patients. A total of 13.3% of the patients had distant metastatic disease, and 10% of the patients had liver metastases, which was higher than the < 5% reported in pediatric cases [17]. A total of 42.1% (8/19) of patients had relapses or metastases after radical surgery, which was also higher than the recurrence rate of 13–25% in pediatric patients [18]. The 5-year OS was 77% in our series, which was worse than the reported data of 98.1% in the EpSSG study [17]. Adult IMT cases appear to be more aggressive, with a higher incidence of recurrence, distant metastases, liver metastases, and worse prognosis.
ALK is a transmembrane tyrosine kinase [19]. The expression and/or rearrangement of ALK in IMT is approximately 50%–60%. ALK expression is associated with ALK rearrangements. The ALK positivity rate was 81.5% (22/27) in this study. Our study showed that adult patients seemed to have a higher rate of ALK rearrangement. In our study, patients with ALK-negative tumors were older than those with ALK-positive tumors (median age, 50 vs. 30 years). Coffin et al. [3] also found that ALK-negative IMT patients were older and had more aggressive tumors in a 59 patients’ analysis.
In 2010, Butrynski et al. [20] first reported the case of an adult IMT patient with ALK gene translocation whose tumor showed significant shrinkage after treatment with crizotinib. There were several trials investigating the efficacy of ALK inhibitors in locally advanced or metastatic IMT (S1 Table). In a phase I/II study, Mosse et al. [14] enrolled 14 pediatric patients with metastatic or inoperable IMT (median age, 7 years; range, 2.0 to 13.5 years) with ALK gene fusion. The ORR and CR rates were 86% and 36%, respectively. The most common drug-related adverse reaction was neutropenia (43%) [14]. The EORTC CREATE trial enrolled 20 patients with advanced/metastatic IMT who received oral crizotinib 250 mg twice daily. The results showed that the ORRs were 66.7% and 14.3%, and the median PFS values were 18.0 months and 14.3 months in ALK-positive patients and ALK-negative patients, respectively [21]. Our study showed that adult IMT patients were also sensitive to crizotinib treatment (ORR 81.3%, CR 18.8%, DCR 87.5%, median progression-free survival 20.8 months).
The mechanisms of crizotinib resistance and subsequent treatment in IMT patients remain unknown. In 2016, Mansfield et al. [22] reported a case of crizotinib-resistant IMT. One patient with IMT who received ceritinib treatment showed significant tumor shrinkage. In a phase I trial, ceritinib showed promising antitumor activity (ORR 70%) and a manageable safety profile in pediatric patients with ALK-positive refractory or recurrent IMT [15]. One patient with TFG-ROS1 fusion-positive IMT of the chest wall with brain metastasis that was refractory to first-and second-generation ROS1 inhibitors showed a rapid response to lorlatinib [23]. In our study, two patients who progressed after crizotinib treatment were treated with ceritinib with PR. One patient who progressed after ceritinib was administered with lorlatinib with PR. This is consistent with the finding that a next generation of ALK inhibitors could be used in crizotinib-resistant ALK-positive IMT patients.
Various ALK partners have been reported in patients with IMT. In our study, IGFBP5-ALK rearrangement was detected using NGS in one patient with uterine IMT. The patient received crizotinib treatment and achieved PR. Only one study has reported IGFBP5-ALK rearrangement in uterine IMTs [24]. IGFBP5 is an insulin growth factor (IGF) protein that regulates the growth-promoting effects of IGFs. Our study showed for the first time that an ALK inhibitor was effective in patients with IGFBP5-ALK rearrangement.
EIMS, first reported by Marino-Enriquez et al. in 2011 [25], is a rare subtype of IMT. As the name suggests, the tumor cells showed obvious epithelioidy and marked heteromorphism. EIMS has a characteristic RANBP2-ALK gene fusion, and the ALK protein is usually positive. EIMS predominantly occurs in the abdominopelvic region, pulmonary EIMS with multiple bone metastases, and ovarian EIMS has also been reported [26,27]. Compared to typical IMT, the clinicopathological features of EIMS are unique, with a high degree of invasion and malignancy [25,28–30]. The seven patients with EIMS in this study originated from the abdominal cavity. They had a median age of 26 years, which is younger than other adult IMT patients, with a female-to-male ratio of 4:3. Tumors cannot be radically resected because of the extensive lesions in all patients. The ALK positivity rate was 100%. All seven patients received crizotinib treatment, with an ORR of 72%. Patients with EIMS were associated with more aggressive tumors; however, the prognosis was similar to that of non-EIMS patients after treatment with an ALK inhibitor in our series.
MPC was observed in four (13.3%) patients in this study. The second primary tumor type included lung adenocarcinoma with EGFR exon 19 mutations, nasopharyngeal carcinoma, pancreatic adenocarcinoma, thyroid cancer, and cervical cancer. However, the underlying mechanism of MPC in patients was unknown. MPC has been reported in other series of IMT [17]. This reminds us to be vigilant about the occurrence of a second primary tumor during the follow-up of patients with IMT.
This study had some limitations. It was a single-center retrospective study. The characteristics of these patients may not be representative of all adult IMT patients. The mechanisms underlying ALK inhibitor resistance have not yet been identified. The sample size of this study was relatively small. However, given the rarity of IMT in adults, it is almost impossible to conduct prospective clinical trials.
The results of this study suggest that adult IMT cases appear to be more aggressive, with a higher incidence of recurrence and metastasis. Also, ALK rearrangements are a common genetic abnormality. Crizotinib showed a high ORR and durable response in patients with advanced adult IMT. A next generation of ALK inhibitors could be used as treatment options for patients with crizotinib-resistant, ALK-positive IMT. The role of the ALK signaling pathway in adult IMT and the mechanism of drug resistance to ALK inhibitors are worthy of further study from clinical and basic aspects.
 XML Download
XML Download