

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Leptin is a 16-kDa fat-derived hormone mainly secreted from white adipose tissue. Leptin controls adipose tissue levels by regulating both food intake and energy expenditure [1,2]. Leptin signaling occurs via leptin receptor (LEPR), which belongs to the cytokine class 1 receptor family located on the plasma membrane [3]. Although there are multiple isoforms of LEPRs (LEPRa-LEPRe), only the longest isoform, LEPRb, has a cytoplasmic portion that associates with the Janus kinase 2 pathway [4]. Binding of leptin to LEPRb phosphorylates specific tyrosine residues on its receptor, which in turn recruits and phosphorylates other mediators such as signal transducer and activator of transcription 3 (STAT3) [5,6]. Phosphorylation of STAT3 induces its dimerization and translocation to the nucleus to activate transcription of its target genes. LEPRb is highly expressed in the hypothalamus [7], as well as in some peripheral tissues, including the lung, adipose tissues, and liver, where it is directly activated by leptin [8–12].
There are three isoforms of small ubiquitin-like modifiers (SUMOs), SUMO1–3, which bind to lysine residues of various proteins. SUMO modification regulates various cellular processes, including signal transduction and gene expression [13]. De-conjugation of SUMO from target proteins is stimulated by SUMO-specific proteases (SENPs). In humans, six isoforms of SENP, SENP1, 2, 3, 5, 6, and 7, have been identified [14,15]. In a previous study, we showed that SENP2 is involved in palmitate-induced fatty acid oxidation (FAO) in myotubes. SENP2 increases upon palmitate treatment and induces the deSUMOylation of peroxisome proliferator-activated receptor δ/γ (PPARδ/γ). DeSUMOylation of PPARδ/γ increases its binding to the promoters of FAO-associated genes, carnitine palmitoyl transferase 1b (Cpt1b) and long-chain acyl-coenzyme A synthetase 1 (Acsl1). As a result, transcription of these FAO-associated enzymes increases in skeletal muscle [16].
Within 15 minutes after leptin treatment in skeletal muscle, leptin stimulates FAO via activation of adenosine monophosphate-activated protein kinase (AMPK) and subsequent inhibition of acetyl-coenzyme A carboxylase [2,17]. Our previous study showed that leptin increases FAO acutely by AMPK activation and chronically by STAT3–SENP2 pathway in the C2C12 myotubes [18]. Leptin binding to LEPRb stimulates STAT3 phosphorylation, which leads to increased Senp2 expression. SENP2 mediates a prolonged FAO increase by enhancing Cpt1b and Acsl1 expression in skeletal muscle [18].
Adipose tissue itself is also a target of leptin [19]. Leptin regulates lipid metabolism in adipose tissue both directly by binding to LEPRb and indirectly via sympathetic innervation [9,20]. Leptin-induced lipolysis in adipose tissue is mainly mediated by sympathetic neuro-adipose connections [21]. Intraperitoneal administration of leptin increases phosphorylation of STAT3 in white adipose tissue and suppresses expression of lipogenic enzymes, including adenosine triphosphate-citrate lyase (ACL) and fatty acid synthase (FAS), in adipose tissues [12]. Moreover, LEPRb knockdown or reconstitution in adipose tissues alters body weight and glucose metabolism [22]. A study showed that leptin directly stimulates FAO but inhibits de novo fatty acid synthesis in rat white adipocytes [23]. In addition, leptin directly decreases FAS expression and increases expression of FAO-associated enzymes, CPT1 and acyl-coenzyme A oxidase (ACO), in isolated white adipocytes [24]. However, the downstream mechanism of leptin/LEPRb binding to regulate fatty acid metabolism in white adipocytes remains unknown.
In this study, we investigated the role of SENP2 in the regulation of fatty acid metabolism by leptin in white adipocytes. First, we tested whether leptin-induced FAO was achieved via AMPK acutely and via SENP2 chronically in 3T3-L1 adipocytes as previously observed in C2C12 myotubes. We confirmed leptin-induced FAO via SENP2 in white adipose tissues in vivo with an adipocyte-specific Senp2 knockout (Senp2-aKO) mouse model. In addition, we examined the involvement of PPAR isoforms in the leptin-SENP2 pathway to regulate FAO in 3T3-L1 adipocytes.
METHODS
Materials, antibodies, and plasmids
Leptin and compound C were purchased from Sigma-Aldrich (St. Louis, MO, USA) and Tokyo Chemical Industry (Tokyo, Japan), respectively. Small interfering RNAs (siRNAs) against STAT3 and SENP2 (Dharmacon, Chicago, IL, USA) and nonspecific siRNA (siNS, negative control, BIONEER, Daejeon, Korea) were used. The specific antibodies used for Western blotting were phospho-STAT3, STAT3 (Cell Signaling Technology, Beverly, MA, USA), PPARα, PPARδ, PPARγ (Santa Cruz Biotechnology, Dallas, TX, USA), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH, Merck, Darmstadt, Germany). The mouse Cpt1b promoter region from −1556 to 53 bp (from the transcription start site) was ligated to the pGL2-basic to generate mCpt1b(−1,556)-luc. The mouse Cpt1b promoter region from −320 to 53 bp was ligated to the pGL2-basic to generate mCpt1b(−320)-luc. The PPAR response element (PPRE) between −247 and −234 bp in the mCpt1b promoter (5′-TGACC-TTTTCCCT-3′) was mutated to 5′-TCTGCTTTTCCCT-3′ in the mCpt1b(−320)mt-luc. The mouse Acsl1 promoter-luciferase reporter vectors, mAcsl1(−1,051)-luc and mAcsl1(−1,051)mt-luc, were described previously [18].
Cell culture, leptin treatment, and siRNA transfection
3T3-L1 preadipocytes were maintained in high glucose Dulbecco’s modified eagle’s medium (DMEM) supplemented with 10% calf serum (Thermo Fisher Scientific, Waltham, MA, USA). Two days after 100% cell confluency, differentiation was initiated by addition of dexamethasone, 3-isobutyl-1-methylxanthine, and insulin in DMEM supplemented with 10% fetal bovine serum (FBS) (day 0). Culture medium was replaced with DMEM (insulin and 10% FBS) at day 2 and then with DMEM (10% FBS) every other day. On day 7, adipocytes were serum starved for 24 hours and treated with 50 ng/mL of leptin on day 8. For siRNA transfection, fully differentiated 3T3-L1 adipocytes were transfected with siRNA against Stat3 (siStat3, 100 nM) or Senp2 (siSenp2, 200 nM) using RNAiMAX (Invitrogen, Thermo Fisher Scientific) for 24 hours. For AMPK inhibition, adipocytes were treated with compound C (10 μM) 1 hour before the leptin treatment.
Animals
Senp2-aKO mice were generated by crossing Senp2-floxed mice with adiponectin-Cre transgenic mice (Jackson lab, Farmington, CT, USA) as described previously [25]. Mice were handled in compliance with the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health, and this study was approved by the Institutional Animal Care and Use Committee of Clinical Research Institute, Seoul National University Bundang Hospital, Republic of Korea (Permit Number: BA-1805-248-044). Control (Senp2-floxed) and Senp2-aKO mice (10-week-old mice) were injected intraperitoneally with recombinant murine leptin (3 mg/kg body weight) dissolved in saline after a 4-hour morning fast. White adipose tissues and soleus muscle were isolated 24 hours after the injection.
Measurement of FAO rate
3T3-L1 adipocytes or mouse tissues were homogenized in ice-cold mitochondria isolation buffer (250 mM sucrose, 10 mM Tris-HCl, and 1 mM EDTA). The lysates were incubated for 2 hours with 0.2 mM [1-14C] palmitate. 14CO2 and 14C-labeled acid-soluble metabolites were quantified using a liquid scintillation counter. Each radioactivity level was normalized to the protein amount of each lysate.
RNA preparation and real-time quantitative polymerase chain reaction
Total RNAs were isolated using TRIzol (Thermo Fisher Scientific) according to the manufacturer’s instructions. Real-time quantitative polymerase chain reaction (RT-qPCR) was performed in duplicate using the SYBR master mix (Takara, Otsu, Japan) and the ABI 7500 Real-time PCR system (Applied Biosystem, Foster City, CA, USA). GAPDH served as the loading control. Each cycle threshold (Ct) value was subtracted from the Ct value of GAPDH (ΔCt) and then subtracted from the value of each control set (ΔΔCt). Relative mRNA levels were expressed as 2−ΔΔCt. Primer sequences are listed in Supplementary Table 1.
Western blot analysis
Cell lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer supplemented with protease inhibitors and subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis. The proteins on the SDS–polyacrylamide gel were transferred onto a nitrocellulose membrane (Whatman, Clifton, NJ, USA). The bands were visualized using Enhanced Chemiluminescence (Pierce, Rockford, IL, USA) with Amersham Imager 680 Blot and Gel imagers (GE Healthcare Life Sciences, Marlborough, MA, USA).
Plasmid transfection and luciferase activity assay
Upon 90% confluency, 3T3-L1 preadipocytes were transfected with pRSV-β-gal (100 ng) and one of the following plasmids: mCpt1b(−1,556)-luc, mCpt1b(−320)-luc, mCpt1b(−320)mt-luc, mAcsl1(−1,051)-luc, or mAcsl1(−1,051)mt-luc (300 ng). Then, the cells were treated with leptin (50 ng/mL) for 24 hours and subjected to luciferase activity assays. Reporter lysis buffer (Promega Corporation, Madison, WI, USA) was used for cell lysis, and the cell extract was mixed with 20 μL of luciferase assay substrate in duplicate. Luciferase activity was measured by using a Lumat LB 9507 machine (Berthold Technologies, Bad Wildbad, Germany), and its relative light unit was normalized by β-galactosidase activity.
Chromatin immunoprecipitation coupled with qPCR
After 24 hours of serum starvation, 3T3-L1 adipocytes were treated with 50 ng/mL of leptin for 24 hours. After crosslinking and DNA fragmentation, immunoprecipitation was performed with antibodies against PPARα, PPARδ, PPARγ, and STAT3. PCR was performed with primers flanking the PPREs in the Cpt1b promoter and the Acsl1 promoter or the STAT3 binding region (around −160 bp) in the Cpt1b promoter. Primer sequences for PCR are indicated in Supplementary Table 1.
Statistical analysis
Statistical analysis was performed in GraphPad Prism version 8 and SPSS Statistics version 26 (IBM Co., Armonk, NY, USA). Statistical significance was assessed using Student’s t-test and two-way analysis of variance (ANOVA). P values below 0.05 were considered statistically significant. The n value in the figure legends means the number of individual experiments.
RESULTS
Leptin increased FAO acutely via AMPK in 3T3-L1 adipocytes
We compared expression of the longest isoform of the LEPR, LEPRb, in different types of mouse tissue using RT-qPCR. The mRNA level of Leprb in subcutaneous adipose tissue (SAT) was comparable to that in soleus muscle, whereas the level in visceral adipose tissue (VAT) was lower (Fig. 1A). Consistently, the mRNA level of Leprb in 3T3-L1 adipocytes was lower than that of C2C12 myotubes (Fig. 1B). Our previous work showed that leptin increases FAO by two different pathways in myotubes; AMPK activation is mainly involved in acute (for example, 6 hours) FAO increase, and prolonged (24 hours) FAO increase is achieved by SENP2-mediated FAO-associated enzyme expression [18]. Therefore, we examined FAO rates in response to leptin treatment after 6 or 24 hours in 3T3-L1 adipocytes. FAO increased by 1.8-fold both 6 and 24 hours after the leptin treatment (Fig. 1C and D). To test whether AMPK activation is involved in the FAO increase induced by short- or long-term leptin treatment, compound C, an AMPK inhibitor, was used for pretreatment 1 hour before the leptin treatments. The FAO increase upon 6 hours of leptin treatment, but not upon 24 hours of leptin treatment, was dramatically reduced (90% reduction) by compound C pretreatment (Fig. 1C and D). These results suggest that AMPK activation is crucial for the acute increase of FAO by leptin in 3T3-L1 adipocytes.

Fig. 1
Leptin increased fatty acid oxidation (FAO) acutely via adenosine monophosphate-activated protein kinase (AMPK) in 3T3-L1 adipocytes. (A) Relative leptin receptor b (Leprb) mRNA levels of several types of fat, brown adipose tissue (BAT), subcutaneous adipose tissue (SAT), visceral adipose tissue (VAT), hypothalamus (Hypothal), soleus muscle, and liver from wild type C57BL/6J mice. The mRNA levels of Leprb were expressed as ΔCt (Leprb Ct - glyceraldehyde-3-phosphate dehydrogenase [Gapdh] Ct). Data are presented as mean±standard error of the mean (SEM) of four different mice. (B) The Leprb mRNA levels of C2C12 myotubes and 3T3-L1 adipocytes were measured and expressed as ΔCt (Leprb Ct - Gapdh Ct). Data are presented as mean±SEM of five to eight different cell preps. (C, D) After pre-treatment with compound C (10 μM) for 1 hour, 3T3-L1 adipocytes were treated with leptin (50 ng/mL) for 6 hours (C) or 24 hours (D), and then FAO was measured. FAO levels were expressed as dpm/μg of proteins (n=3). All data are presented as mean±SEM. n, number of individual experiments. aP<0.05 vs. vehicle, bP<0.05 vs. leptin without compound C.
![]()
Leptin increased Senp2 expression via STAT3 in 3T3-L1 adipocytes
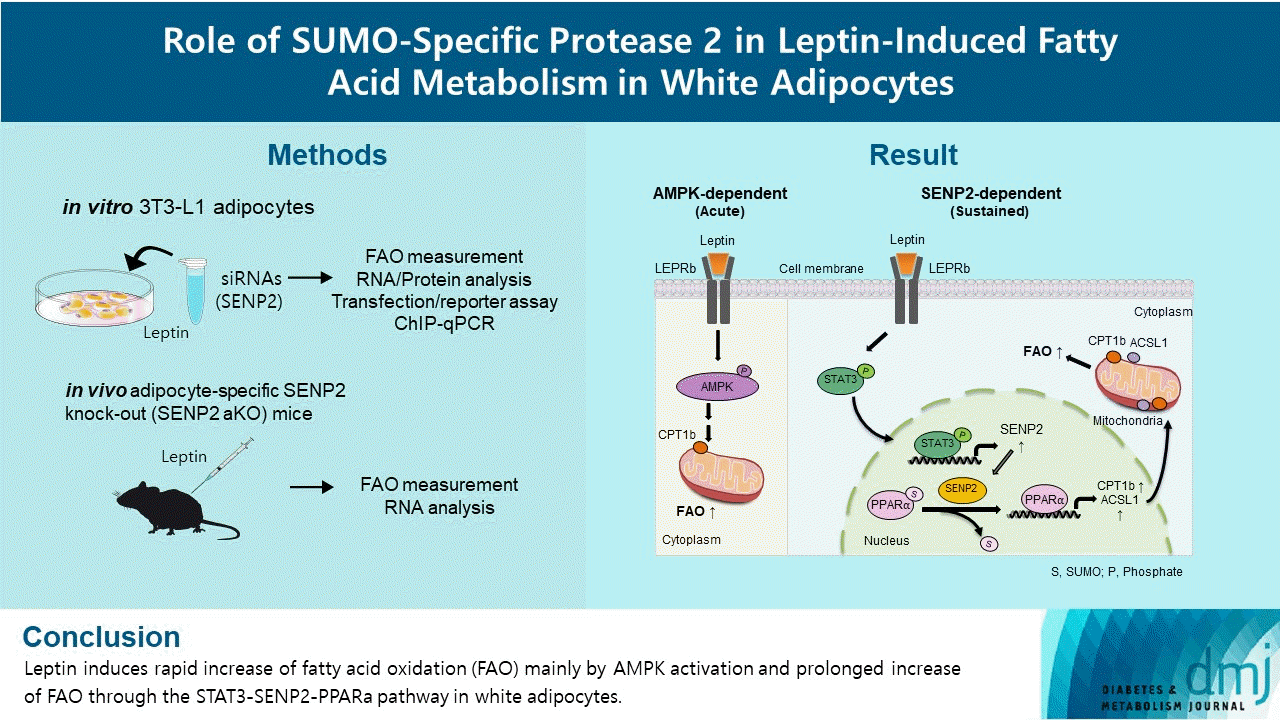
Our previous study demonstrated that Senp2 expression increases through the STAT3 signaling pathway upon leptin treatment, which plays an important role in chronic induction of FAO by leptin in C2C12 myotubes [18]. We tested whether STAT3 has a similar role in 3T3-L1 adipocytes. In fully differentiated 3T3-L1 adipocytes, the level of phospho-STAT3 increased by leptin treatment in a dose-dependent manner (Fig. 2A). Senp2 mRNAs increased gradually until 24 hours after leptin treatment (Fig. 2B). When STAT3 was knocked down by siStat3 before leptin treatment (Supplementary Fig. 1), Senp2 mRNA level was not increased by leptin (Fig. 2C). These results suggest that leptin increases Senp2 expression via STAT3 in 3T3-L1 adipocytes.
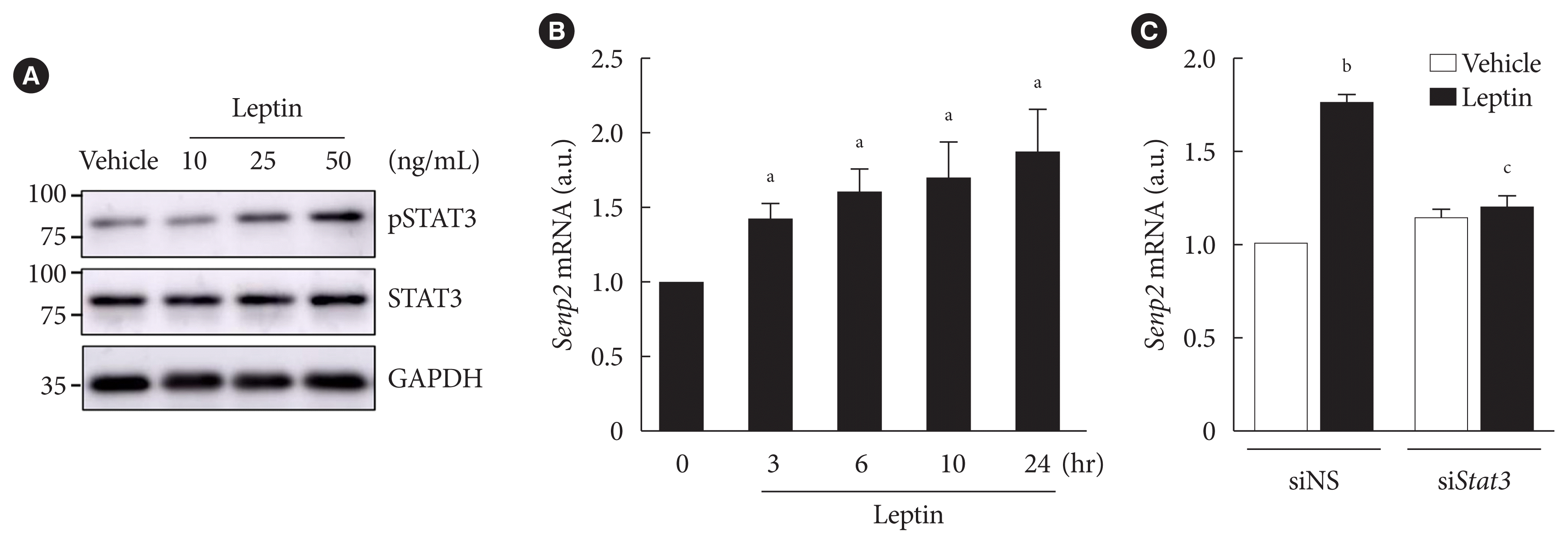
Fig. 2
Leptin increased SUMO-specific protease 2 (Senp2) expression via signal transducer and activator of transcription 3 (STAT3) in 3T3-L1 adipocytes. (A) After serum starvation for 24 hours, 3T3-L1 adipocytes were treated with leptin at different concentrations (10, 25, or 50 ng/mL) for 3 hours. Cell lysates were subjected to Western blotting using antibodies against phospho-STAT3 (pSTAT3) and total STAT3 (STAT3). (B) After serum starvation for 24 hours, 3T3-L1 adipocytes were treated with leptin (50 ng/mL) for 3, 6, 10, or 24 hours. Real-time quantitative polymerase chain reaction analysis was performed with Senp2 primers. The mRNA level of vehicle was expressed as 1, and the others were expressed as its relative values (n=5). (C) After nonspecific small interfering RNA (siNS) or siRNA against Stat3 (siStat3) (100 nM) treatment for 48 hours, 3T3-L1 adipocytes were treated with leptin (50 ng/mL) for 24 hours. The Senp2 mRNA level of siNS without leptin treatment was expressed as 1, and the others were expressed as its relative values (n=3). All data are presented as mean±standard error of the mean. GAPDH, glyceraldehyde-3-phosphate dehydrogenase; a.u., arbitrary unit. aP<0.05 vs. 0 hour, bP<0.05 vs. vehicle, cP<0.05 vs. siNS/leptin.
![]()
Leptin increased expressions of FAO-associated enzymes through SENP2 in 3T3-L1 adipocytes
Next, we investigated whether leptin increased expression of FAO-associated enzymes, such as Cpt1b and Acsl1, through SENP2 in adipocytes. Cpt1b mRNA level rapidly increased only 3 hours following leptin treatment, whereas Acsl1 mRNA did not increase until 10 hours following leptin treatment (Fig. 3A). The mRNA levels of Cpt1b and Acsl1 both reached their peaks 24 hours after leptin treatment (Fig. 3A). When SENP2 was knocked down with siSenp2 (Fig. 3B), the leptin-induced increase of Cpt1b and Acsl1 mRNA levels was suppressed by 80% to 90% (Fig. 3C). These results suggest that leptin-induced increases in Cpt1b and Acsl1 expression were mainly mediated by SENP2 in adipocytes. Because previous reports indicated that leptin increases expression of mitochondrial uncoupling protein 2 (Ucp2), Pparα, and Aco but decreases Fas expression in rat white adipocytes [24,26], we examined their mRNA levels after leptin treatment in 3T3-L1 adipocytes. Consistently, leptin treatment increased mRNA levels of Ucp2, Pparα, and Aco (1.3- to 1.5-fold) and decreased Fas mRNA expression (30% reduction) in 3T3-L1 adipocytes (Supplementary Fig. 2). However, the effects of leptin were eliminated by SENP2 knockdown (Supplementary Fig. 2), suggesting that SENP2 was also involved in the regulation of Ucp2, Pparα, Aco, and Fas expression by leptin in 3T3-L1 adipocytes.
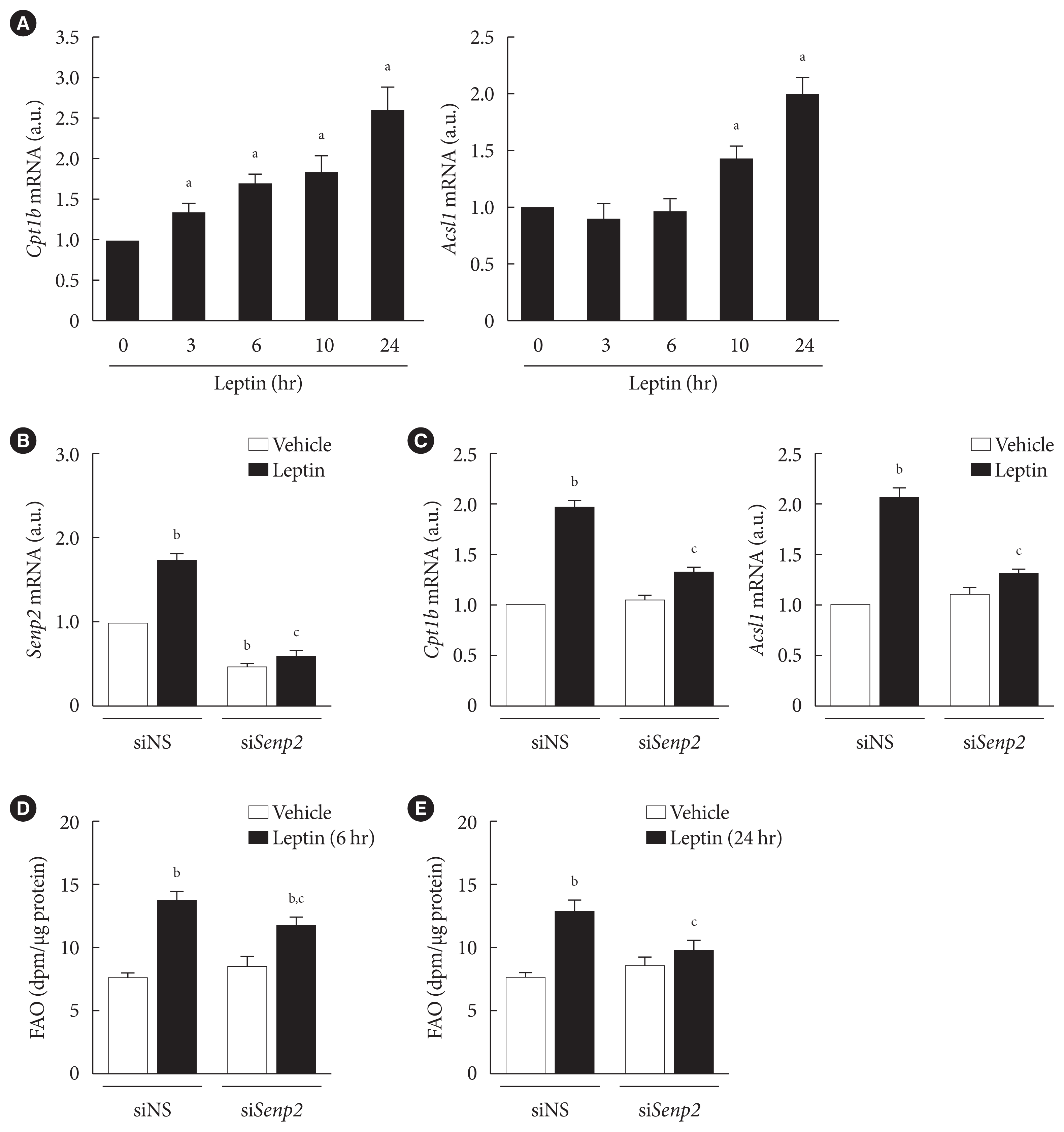
Fig. 3
Leptin increased expressions of fatty acid oxidation (FAO)-associated enzymes through SUMO-specific protease 2 (SENP2) in 3T3-L1 adipocytes. (A) After serum starvation for 24 hours, 3T3-L1 adipocytes were treated with leptin (50 ng/mL), and real-time quantitative polymerase chain reaction (RT-qPCR) analysis was performed using primers specific to carnitine palmitoyl transferase 1b (Cpt1b, left) or long-chain acyl-coenzyme A synthetase 1 (Acsl1, right). The mRNA level at 0 hour was expressed as 1, and the others were expressed as its relative values (n=5). (B, C) After nonspecific small interfering RNA (siNS) or siRNA against Senp2 (siSenp2, 200 nM) treatment for 48 hours, 3T3-L1 adipocytes were treated with leptin for 24 hours, and then RT-qPCR analysis was performed using primers for Senp2 (B), Cpt1b and Acsl1 (C). The mRNA levels of siNS without leptin treatment were expressed as 1, and the others were expressed as their relative values (n=3). (D, E) 3T3-L1 adipocytes were transfected with siNS or siSenp2 for 48 hours followed by leptin treatment for 6 hours (D) or 24 hours (E), and then FAO levels of the adipocytes were measured (n=3). All data are presented as mean±standard error of the mean. a.u., arbitrary unit. aP<0.05 vs. 0 hour, bP<0.05 vs. siNS/vehicle, cP<0.05 vs. siNS/leptin.
![]()
We also examined the effect of SENP2 knockdown on leptin-induced FAO in 3T3-L1 adipocytes. Although SENP2 knockdown suppressed the increase in FAO 6 hours after leptin treatment by only 30% (Fig. 3D), the increase in FAO 24 hours following leptin treatment was suppressed by 80% (Fig. 3E). Therefore, these results reveal that the chronic increase of FAO by leptin occurs mainly via SENP2-mediated expression of FAO-associated enzymes in 3T3-L1 adipocytes.
Leptin increased FAO in white adipose tissues in vivo via SENP2
Next, we confirmed the role of SENP2 in leptin-induced adipocyte FAO in wild type (control) and Senp2-aKO mice. Control and Senp2-aKO mice were injected with leptin or saline intraperitoneally, and adipose tissues, including VAT and SAT, and soleus muscle were isolated 24 hours after the injection. Although 1.8-fold increases in FAO were observed in the adipose tissues of control mice following leptin injection, leptin-injected Senp2-aKO mice had no increase of FAO in both VAT and SAT (Fig. 4A). On the other hand, soleus muscle of both control and Senp2-aKO mice showed more than 2-fold increases in FAO by leptin. These results indicate that SENP2 mediated the leptin-induced increase in FAO in white adipose tissues. In addition, expression of SENP2 and FAO-associated enzymes were measured in adipose tissues and muscle. Adipose tissue-specific Senp2 mRNA reduction was confirmed in Senp2-aKO mice (Fig. 4B, top panel). The mRNA levels of Senp2, Cpt1b, and Acsl1 increased around 2-fold in adipose tissues and soleus muscle of control mice following leptin treatment (Fig. 4B). In contrast, leptin treatment failed to increase Cpt1b and Acsl1 mRNAs in adipose tissues of Senp2-aKO mice; expression of these enzymes only increased in soleus muscle (Fig. 4B, middle and bottom panels). The in vivo study strongly demonstrates that SENP2 was responsible for the leptin-induced chronic increase of FAO through transcriptional induction of FAO-associated enzymes in adipose tissues.
Fig. 4
Leptin increased fatty acid oxidation (FAO) in white adipose tissues in vivo via SUMO-specific protease 2 (SENP2). (A, B) Ten weeks old control and adipocyte-specific Senp2 knockout (Senp2-aKO) mice were injected with leptin (3 mg/kg) or saline intraperitoneally. Mice tissues, including visceral adipose tissue (VAT), subcutaneous adipose tissue (SAT), and soleus muscle (Soleus), were obtained 24 hours after the injection, and FAO (A) or mRNA levels of Senp2, carnitine palmitoyl transferase 1b (Cpt1b), and long-chain acyl-coenzyme A synthetase 1 (Acsl1) (B) were measured. (A) FAO was expressed as dpm/μg of proteins (n=3 mice). (B) The mRNA level of each tissue from saline-injected control mice was expressed as 1, and the others were expressed as its relative values (n=3 mice). All data are presented as mean±standard error of the mean. a.u., arbitrary unit. aP<0.05 vs. control/saline, bP<0.05 vs. control/leptin.
![]()
SENP2 increased binding of PPARα on PPRE sites in the Cpt1b and Acsl1 promoters upon leptin treatment in 3T3-L1 adipocytes
Because SENP2 increases Cpt1b and Acsl1 transcription by increasing PPARδ and PPARγ binding to the PPREs of the Cpt1b and Acsl1 promoters in C2C12 myotubes [16], we measured the effect of leptin on Cpt1b and Acsl1 promoter activities in 3T3-L1 preadipocytes. Leptin treatment led to a 1.5-fold increase in the luciferase activity of the cells transfected with mCpt1b(−1,556)-luc or with mCpt1b(−320)-luc but not with mCpt1b(−320)mt-luc containing mutations at a potential PPRE site (Fig. 5A). Likewise, leptin treatment led to an almost 2-fold increase in the promoter activity of mAcsl1(−1,051)-luc but not of mAcsl1(−1,051)mt-luc containing mutations at a PPRE site (Fig. 5B). These results revealed that PPAR binding on the PPRE sites in the Cpt1b and Acsl1 promoters is necessary for the increase of Cpt1b and Acsl1 mRNAs upon leptin treatment in 3T3-L1 adipocytes.
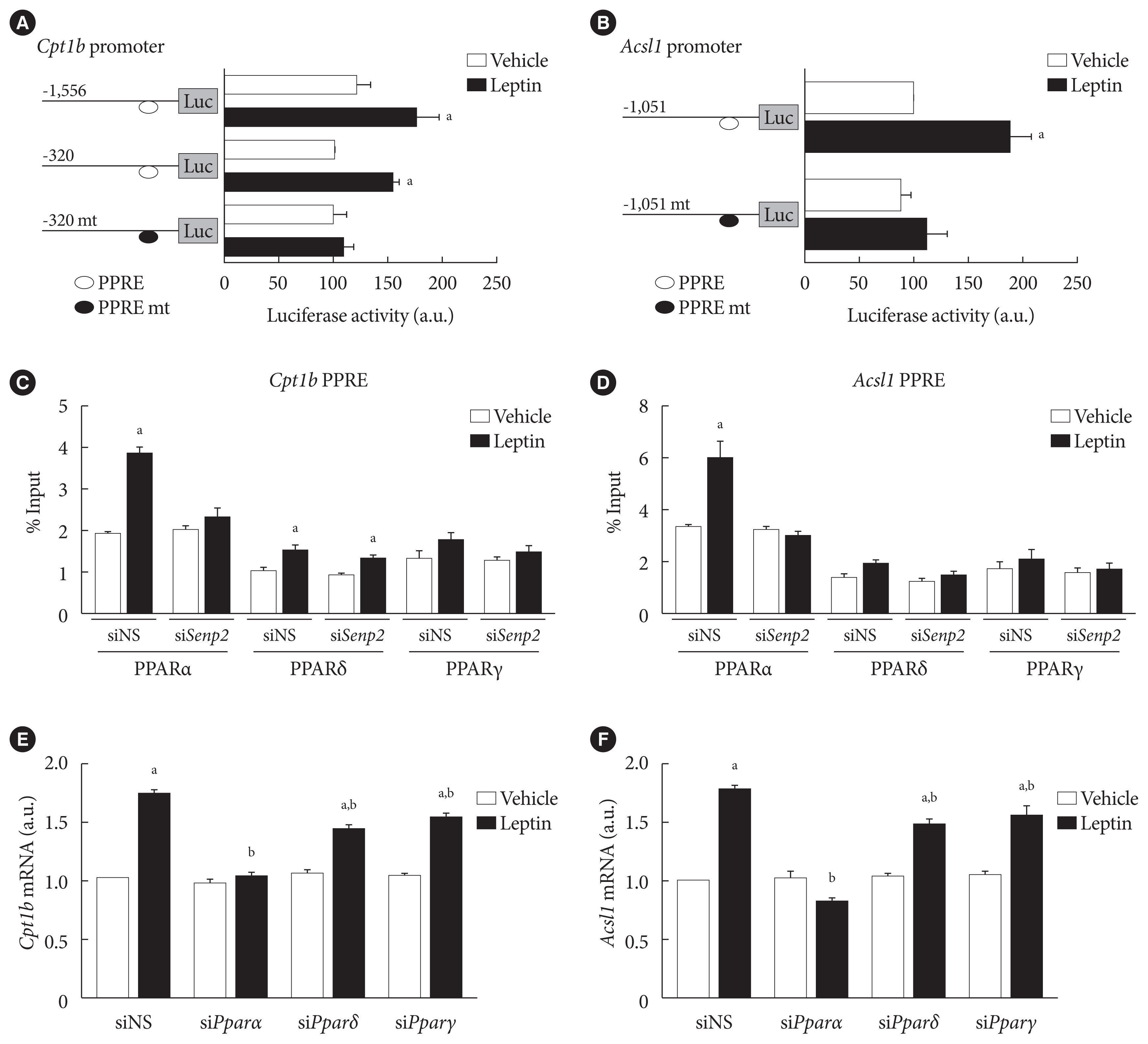
Fig. 5
SUMO-specific protease 2 (SENP2) increased binding of peroxisome proliferator-activated receptor α (PPARα) on PPAR response element (PPRE) sites in the carnitine palmitoyl transferase 1b (Cpt1b) and long-chain acyl-coenzyme A synthetase 1 (Acsl1) promoters upon leptin treatment in 3T3-L1 adipocytes. (A, B) 3T3-L1 preadipocytes were transfected with 300 ng of the Cpt1b promoter-luc constructs (A), and the Acsl1 promoter-luc constructs (B), followed by leptin treatment (50 ng/mL) for 24 hours. Luciferase activity of the cells transfected with mCpt1b(−320)-luc or mAcsl1(−1,051)-luc without leptin treatment was expressed as 100, and the others were expressed as their relative values (n=5). (C, D) After nonspecific small interfering RNA (siNS) or siRNA against Senp2 (siSenp2, 200 nM) treatment for 48 hours, 3T3-L1 adipocytes were treated with leptin for 24 hours, and then subjected to chromatin immunoprecipitation (ChIP)-coupled quantitative polymerase chain reaction analysis using antibodies against PPARα, PPARδ, and PPARγ and the Cpt1b PPRE primers (C) or Acsl1 PPRE primers (D). Binding activity was expressed as its % input (n=3). (E, F) After siNS, siPparα, siPparδ, or siPparγ (100 nM) treatment for 48 hours, 3T3-L1 adipocytes were treated with leptin for 24 hours, and then subjected to real-time quantitative polymerase chain reaction analysis using Cpt1b primers (E) or Acsl1 primers (F). The mRNA levels of siNS/vehicle were expressed as 1, and the others were expressed as their relative values (n=4). All data are presented as mean±standard error of the mean. a.u., arbitrary unit. aP<0.05 vs. vehicle, bP<0.05 vs. siNS/leptin.
![]()
To determine whether leptin affects binding affinities of PPAR isoforms to the PPREs of Cpt1b and Acsl1 promoters, chromatin immunoprecipitation (ChIP) was performed using PPARα, PPARδ, and PPARγ antibodies. Leptin treatment led to 2- and 1.6-fold increases in the binding of PPARα and PPARδ on the PPRE of the Cpt1b promoter, respectively (Fig. 5C). Similarly, leptin increased binding of PPARα (1.9-fold), but not of PPARδ or PPARγ, to the PPRE of the Acsl1 promoter (Fig. 5D). Upon SENP2 knockdown, leptin-induced PPARα binding to the PPREs of Cpt1b and Acsl1 promoters disappeared (Fig. 5C and D). These results suggested that PPARα was important for leptin-induced Cpt1b and Acsl1 expression in adipocytes. To confirm whether PPARα is necessary for the leptin-induced expression of Cpt1b and Acsl1 in 3T3-L1 adipocytes, PPAR isoforms were knocked down using specific siRNAs (Supplementary Fig. 3). PPARα knockdown eliminated the increase of Cpt1b and Acsl1 mRNAs induced by leptin, whereas PPARδ and PPARγ knockdown led to only a 20% to 30% reduction (Fig. 5E and F). None of the PPAR isoform knockdowns affected the basal mRNA levels of Cpt1b or Acsl1. To summarize, leptin increased Cpt1b and Acsl1 expression by facilitating binding of PPARs, mostly PPARα, to the PPREs of Cpt1b and Acsl1 promoters in 3T3-L1 adipocytes. This process was mediated by SENP2.
DISCUSSION
In this study, we revealed that SENP2 mainly mediated the chronic induction of FAO by leptin, whereas AMPK regulated acute induction of FAO in 3T3-L1 adipocytes. The effects of leptin in adipocytes were similar to those observed previously in C2C12 myotubes [18]. SENP2-mediated FAO increase was achieved by transcriptional increases in FAO-associated enzymes, such as Cpt1b and Acsl1. These findings were confirmed in vivo with Senp2-aKO mice. Intriguingly, our results indicated that different PPAR isoforms mediated the transcription of Cpt1b and Acsl1 in adipocytes and myotubes. In adipocytes, leptin increased Cpt1b and Acsl1 transcription through SENP2-mediated PPARα binding to the promoters of Cpt1b and Acsl1, whereas leptin increases PPARδ/γ binding in C2C12 myotubes [18].
When Cpt1b and Acsl1 mRNA levels were monitored after leptin treatment, Cpt1b mRNA levels rapidly increased, but the increase in Acsl1 mRNA levels was delayed for 10 hours post-treatment. A study reported that STAT3 directly binds to the Cpt1b promoter and increases transcription of Cpt1b upon leptin treatment in breast cancer stem cells [27]. Therefore, we tested whether leptin-induced phospho-STAT3 directly increased transcription of Cpt1b in 3T3-L1 adipocytes. Using ChIP assays, we found that leptin increased STAT3 binding to the Cpt1b promoter by 1.4-fold (Supplementary Fig. 4A). Based on the result, we conclude that Cpt1b expression was regulated by leptin through both STAT3 directly and the STAT3–SENP2 pathway. This result may explain the different expression timelines for Cpt1b and Acsl1 mRNAs after leptin treatment.
Previous reports indicate that leptin directly and rapidly increases FAO in skeletal muscle by AMPK activation within 15 minutes of leptin treatment; leptin also increases FAO via the sympathetic nervous system-mediated activation of AMPK over a period of several hours [2,17]. Future studies are needed to confirm that leptin acutely increases FAO through AMPK activation in adipose tissues in vivo. In addition, these studies should reveal whether leptin induces the same signaling pathways, both the AMPK activation and the STAT3–SENP2 pathway, in other tissues expressing LEPRb such as the heart and liver.
Our previous report showed that prolonged leptin-stimulated FAO occurs via deSUMOylation of PPARδ/γ by SENP2 in C2C12 myotubes [18]. Although PPARγ expression is predominant in adipocytes, our results revealed that PPARα is mainly responsible for the increase of Cpt1b and Acsl1 expression following leptin treatment in 3T3-L1 adipocytes [28]. Consistent with previous results [29], we confirmed that FAO was increased by WY14643, a PPARα agonist, but not by rosiglitazone, a PPARγ agonist, in 3T3-L1 adipocytes (Supplementary Fig. 4B). It remains unclear why different PPAR isoforms are involved in Cpt1b and Acsl1 expression in adipocytes and myotubes. However, different tissue-specific factors may be involved in recruiting PPAR isoforms to the PPREs of these promoters. In addition, SUMOylation of PPARα is known to inhibit PPARα activity [30]. Therefore, we propose that SENP2 increases PPARα binding to the PPREs of Cpt1b and Acsl1 promoters by direct deSUMOylation of PPARα. However, this possibility should be investigated in further experiments. Interestingly, PPARα agonists increase adipocyte differentiation and FAO, but do not stimulate lipid accumulation in 3T3-L1 adipocytes and human adipocytes [31,32]. In addition, lipogenesis, inflammation, and cholesterol ester accumulation increase in adipose tissues of adipose-specific PPARα knockout mice [33]. These results suggest a pivotal role for PPARα and the regulation of PPARα activity by SENP2 in adipocytes.
Leptin also modestly increases the expressions of Ucp2, Pparα, and Aco, whereas it decreases Fas expression [24,26]. Notably, our study showed that SENP2 was also involved in the effect of leptin on the expressions of these proteins in 3T3-L1 adipocytes (Supplementary Fig. 2). Therefore, future studies should investigate whether leptin also regulates transcription of these proteins, including FAS, through the SENP2–PPARα pathway. In addition, it will be important to determine the role of SENP2 in relation to leptin-induced thermogenesis, where mitochondrial Ucp1 gene expression is increased by leptin in brown adipose tissue [34]. In addition, leptin upregulates adipose triglyceride lipase expression via Janus kinase/STAT and mitogen-activated protein kinase signaling pathways [35]. Therefore, the effects of leptin on additional enzymes related to fatty acid metabolism via SENP2 should be investigated.
To summarize, our findings verify the role of SENP2 in the regulation of fatty acid metabolism by leptin in adipocytes. These results could be used to develop treatments for obesity and type 2 diabetes mellitus.
 XML Download
XML Download