

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Diabetes mellitus is a globally fast-growing chronic metabolic disease with significant social, health, and economic consequences [1, 2]. Accurate information about intrinsic insulin secretion activity and residual islet beta-cell function is essential for the classification and therapeutic monitoring of patients with diabetes mellitus [3, 4]. C-peptide and insulin are generated by proteolytic cleavage of proinsulin and are released into the blood in equimolar amounts [5]. Serum C-peptide and insulin are the most routinely used biomarkers to assess residual beta-cell function in patients with diabetes. Because the kidneys clear C-peptide more slowly than the liver clears insulin, the concentration of C-peptide in the blood is three to five times higher than that of insulin [6, 7], providing a wider test window to estimate fluctuations in beta-cell responses. In patients treated with exogenous insulin, serum C-peptide is a better indicator of beta-cell activity because of cross-activity between endogenous and exogenous insulin.
Various immunoassays are used in laboratories to measure serum C-peptide concentrations. However, we previously revealed that, despite the substantial effort that has been made to standardize serum C-peptide measurements, measurement results from different laboratories remain highly variable in China [8, 9]. Therefore, well-performing methods are required to serve as an accuracy base for routine assays and to improve the agreement among laboratories in China. Isotope dilution liquid chromatography-tandem mass spectrometry (ID-LC–MS/MS) is widely accepted as a reference method for numerous clinical indicators, including C-peptide, owing to its specificity, accuracy, and sensitivity [10, 11]. To improve the harmonization of serum C-peptide measurements and assign the values of control materials in the External Quality Assessment (EQA) program in China, in this study, the Chinese National Center for Clinical Laboratories (NCCL; Beijing, China) established a reliable serum C-peptide ID-LC–MS/MS method based on simple one-step anion-exchange solid-phase extraction (SPE). The ID-LC–MS/MS method was comprehensively and carefully validated and exhibited desirable performance. The ID-LC–MS/MS method and the six most widely used routine immunoassays were compared using split-sample measurement. The ID-LC–MS/MS method will be used to conduct a trueness verification program and to define target values of commutable EQA materials (e.g., serum pools), which will aid in improving the harmonization of C-peptide measurements in China.
MATERIALS AND METHODS
Method development
Chemicals and equipment
C-peptide primary certified reference material (CRM; NMIJ CRM 6901-c with a certified value of 104±5 mg/L) approved by the Joint Committee for Traceability in Laboratory Medicine (JCTLM) was purchased from the National Metrology Institute of Japan (Tsukuba, Japan) [12]. Internal standard (IS), [27, 31-13C3]-C-peptide, with an isotopic purity of 98%, was purchased from GL Biochem (Shanghai, China). Acetonitrile and methanol were purchased from Thermo Fisher Scientific (Waltham, MA, USA). Formic acid and zinc sulfate were purchased from Sigma-Aldrich (Darmstadt, Germany). Ultrapure deionized water (≥18.2 MΩ cm) was prepared using a Milli-Q water purifier (Billerica, MA, USA). A 6500 Plus Triple Quadrupole Mass Spectrometer (AB Sciex, Framingham, MA, USA) coupled with a Waters Acquity UPLC Fl-I Class System (Waters, Milford, MA, USA) was used for LC-MS/MS analysis.
Preparation of calibration solutions
To prepare a primary standard solution of NMIJ CRM 6901-c with a certified value of 104±5 mg/L according to the manufacturer’s instruction, 1±0.01 g deionized water was added into a vial and 1 g of NMIJ CRM 6901-c was accurately weighed on an analytical balance (Mode MSA 125P; Sartorius, Göttingen, Germany), dissolved in 100 g fetal bovine serum (FBS), and added to the vial to prepare a stock standard solution of 898.29±21.55 ng/g (299,427.0±7,186 pmol/L). We ascertained that the FBS did not contain human C-peptide, and no interferences were observed during analysis. A working standard solution with a C-peptide concentration of 10.37 ng/g±0.25 (3,456.6±82.94 pmol/L) was prepared similarly by diluting the stock standard solution in FBS. In the same manner, an IS working standard solution of 27.52 ng/g (9,173.2 pmol/L) was prepared by dissolving isotopically labeled C-peptide in FBS. All standard solutions were gravimetrically prepared, aliquoted to 1 mL, and stored at -80°C.
Quantitation
The bracketing calibration method was used for serum C-peptide quantitation, and serum samples were calibrated using low- and high-concentration standard solutions. The serum sample and standard solutions were spiked with an IS solution so that the C-peptide-to-IS signal ratios for the serum sample and low- and high-concentration standard solutions were approximately 1, 0.9, and 1.1, respectively. The serum C-peptide concentration (C; pmol/L) was calculated as follows:
where ISam, ILow, and IHi are the C-peptide/IS signal ratios of the serum samples, low-concentration standard solution, and high-concentration standard solution, respectively; WLow and WHi are the C-peptide/IS mass ratios of the low- and high-concentration standard solutions, respectively; MIS is the mass of the IS in serum samples; MSer and DS are the mass and density of serum samples, respectively; and MS is the relative molecular mass of C-peptide.
Sample preparation
For each sample, 50 μL of IS solution was accurately weighed and transferred into 2-mL centrifuge tubes. To ensure that the C-peptide-to-IS signal ratio was 1, an appropriate volume (0.05-1.0 mL) of serum was accurately weighed based on the primary result from a routine method and mixed with the IS solution. One milliliter of 0.1 mmol/L ZnSO4 solution (pH=3.5-4.0) was then added to precipitate the proteins in the serum samples. After vortex mixing and centrifugation at 21,100×g, 4°C for 10 min, the supernatants were transferred to Oasis mixed-anion-exchange (MAX) cartridges (Water) preconditioned with 1 mL of methanol and then with 1 mL of water. The loaded cartridges were washed sequentially with 1 mL of 0.1 mmol/L ZnSO4 solution (pH=3.5-4.0) and 1 mL of methanol. Finally, C-peptide was eluted with 2 mL of methanol containing 1% formic acid. The eluate was dried under nitrogen and reconstituted in 100 μL of 10% acetonitrile/water containing 0.4% formic acid for LC-MS/MS analysis.
LC-MS/MS analysis
For chromatographic separation, 30 μL of reconstituted eluate was injected into an XSelect Peptide CSH C18 column (3.5 μm, 2.1 mm×100 mm, Waters), which was maintained at 40°C. Mobile phase A was water containing 0.4% formic acid, and mobile phase B was acetonitrile containing 0.4% formic acid. The following linear gradient with a flow rate of 0.4 mL/min was used: 0 minute, 10% B, 2 minutes, 25% B; 3.5 minutes, 40% B; 5.5 minutes, 40% B; 5.51 minutes, 100% B; 6.5 minutes, 100% B; 6.51 minutes, 10% B; and 8 minutes 10% B.
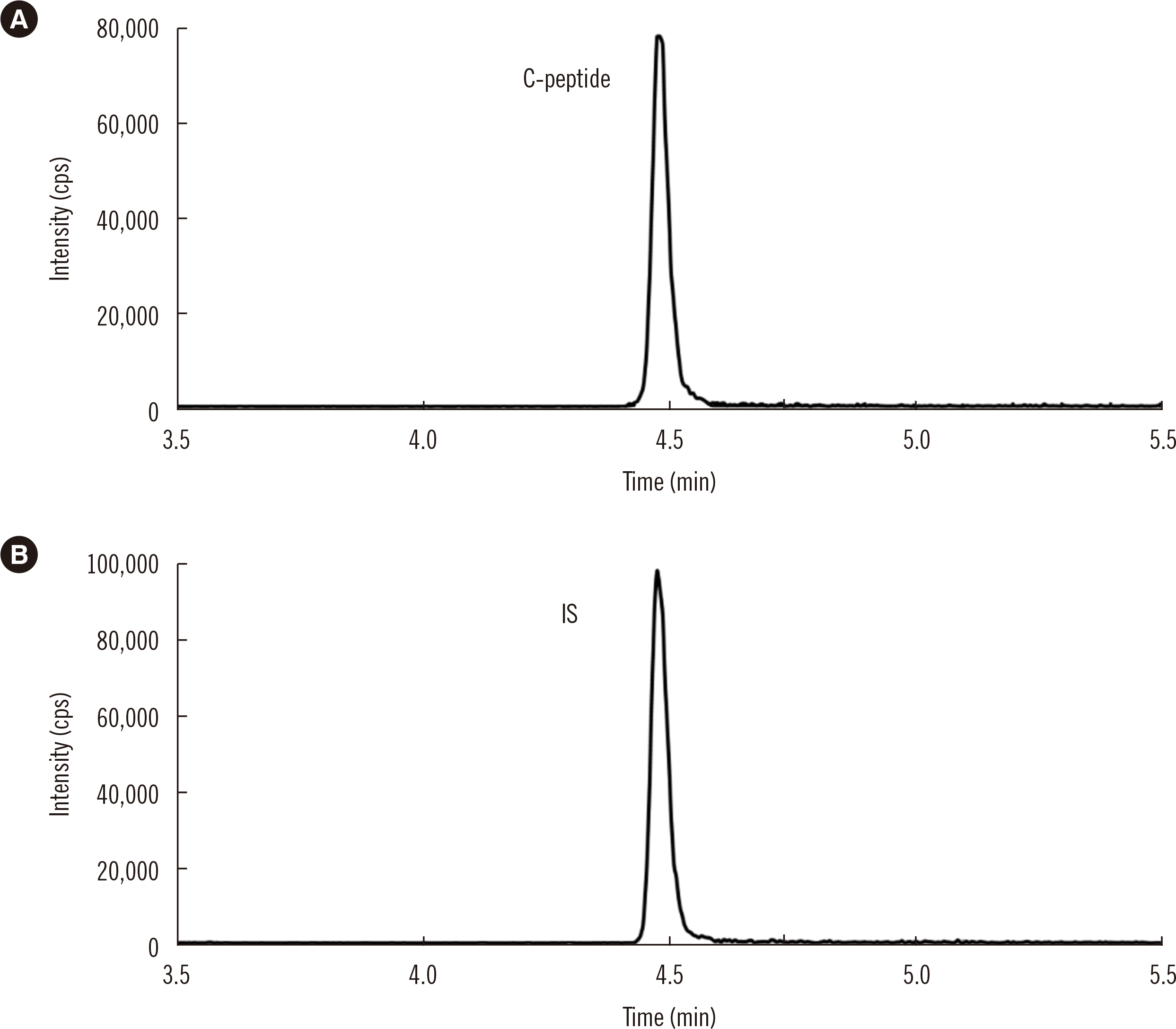
The mass spectrometer was operated in positive electrospray ionization mode with multiple reaction monitoring. Transitions of m/z 1,007.7 (3+) → 147.2 (1+) for C-peptide and m/z 1,011.7 (3+) → 147.2 (1+) for the IS were monitored for quantification under the following conditions: source gas temperature, 400°C; ion spray voltage, 5,500 V; curtain gas, 30 psi; nebulizer gas, 90 psi; auxiliary gas, 60 psi; collision gas, 8 psi; declustering potential, 120 V; entrance potential, 7 V; collision energy, 28 eV; collision exit potential, 12 V; Q1 resolution: low, Q2 resolution: unit. Representative MS/MS chromatograms of C-peptide and IS in a serum sample are shown in Fig. 1. Data were analyzed using the Analyst v.1.7 software (AB Sciex).
Method validation
Absolute recovery of sample preparation, matrix effect, limit of quantification (LOQ), and limit of detection (LOD)
The absolute recovery of sample preparation was determined by adding the same amount of IS to serum samples before and after extraction. The absolute recovery was calculated as the ratio of the peak area of IS added to the serum before extraction to the peak area of IS added to the serum after extraction. Four types of samples—normal, hemolytic, lipemic, and icteric serum samples—were used to evaluate the matrix effect of the developed method. Each extracted matrix and neat standard solution were spiked with a known amount of IS solution. The matrix effect was calculated as the ratio of the peak area of IS added to the extracted matrix to the peak area of IS added to the neat standard solution. The LOQ and LOD were determined using serum samples diluted with FBS and were defined as the lowest analyte concentrations with a signal-to-noise (S/N) ratio of at least 10:1 and 3:1, respectively.
Trueness and analytical recovery
A spiking recovery experiment was used to evaluate the trueness of the ID-LC–MS/MS method. One serum sample (1.86. ng/g [620.0 pmol/L]) was spiked with different amounts of standard solutions to prepare low-, medium-, and high-concentration serum samples, which were measured in duplicate in three runs on three consecutive days. The mean results of the three runs were calculated as the average recoveries.
Imprecision and uncertainty evaluation
The imprecision of the ID-LC–MS/MS method was determined according to CLSI document EP15-A3 [13]. Four serum pool samples with different concentrations of C-peptide (199.3-2,836.8 pmol/L) were measured in triplicate in five runs on five consecutive days. The within- and between-run and total imprecision were calculated using ANOVA.
According to the International Organization for Standardization Guide Uncertainty of Measurement [14], the uncertainty of the results from the ID-LC–MS/MS method was estimated at four levels. Both the uncertainty derived from repeated measurements (type A) and other factors (type B), such as the purity of the reference materials and inaccuracy in weighing, were calculated.
Method comparison
Serum samples and measurement procedure
Seventy-six individual serum samples and five frozen serum pool samples were analyzed in triplicate using the ID-LC–MS/MS method and six routine immunoassays from Roche (Cobas e801, Basel, Switzerland), Abbott (I2000, Chicago, IL, USA), Beckman (Dxi800, Brea, CA, USA), Siemens (ADVIA Centaur XP, Munich, Germany), Mindray (CL8000i, Shenzhen, China), and Snibe (Maglumi X8, Shenzhen, China). All manufacturers claim that their assays are traceable to WHO IRP 84/510, except Beckman, whose assay is traceable to WHO ISR13/146. Detailed information on the immunoassays is presented in Supplemental Data Table S1. We first assessed the imprecision of the routine immunoassays by measuring three serum samples with different concentrations in two replicates per day for three consecutive days; if the imprecision was within the acceptable limit (8%), they were deemed appropriate and used to measure the individual serum samples and frozen serum pool samples. The 76 individual samples (173-3,612 pmol/L) were leftover patient samples collected at Beijing Hospital (Beijing, China) between January 2022 and August 2022. The five frozen serum pool samples (199.3-2,836.8 pmol/L) were prepared by mixing individual samples with similar concentrations. Samples with hemolysis, icterus, or lipemia were considered deviant and excluded. Both individual serum samples and serum pool samples were aliquoted, stored at -80°C, and distributed to laboratories on dry ice. This study was approved by the Ethics Committee of Beijing Hospital (2018BJYYEC-019-01).
Recalibration
The five serum pool samples were used to recalibrate the results of the routine assays for individual serum samples. An ordinary linear regression (OLR) equation between the developed ID-LC–MS/MS method and the routine immunoassays for the five serum pool samples was used to draw new calibration curves for the routine assays. The results of the routine assays for individual serum samples were recalibrated based on the relevant calibration curve.
Statistical analysis
Before the method comparison, outliers were identified and removed using visual detection and the generalized extreme studentized deviate test according to CLSI document EP09-A3 [15]. OLR was used to evaluate the agreement and correlations between the ID-LC–MS/MS methods and the routine assays. Slopes and intercepts with 95% confidence intervals (CIs) and Spearman’s correlation coefficients (R2) were calculated. Bland-Altman (BA) plots were generated to estimate the percentage difference between the developed method and routine assays. The correlation and percentage difference of the assays were re-evaluated after recalibration with serum pools assigned by the ID-LC–MS/MS method. Data were analyzed using Microsoft Excel 2016 (Microsoft, Redmond, WA, USA) and MedCalc statistical software 18.11.6-64-bit (MedCalc Software, Ostend, Belgium). The Kolmogorov-Smirnov test was used to examine whether the data followed a normal distribution.
RESULTS
Absolute recovery of sample preparation, matrix effect, LOQ, and LOD
The absolute recovery of sample preparation for the ID-LC–MS/MS method was 78.2%–84.7%, 80.6%–83.3%, and 79.8%–83.6% at concentrations of 199.3, 634.4, and 1,573.1 pmol/L, respectively (N=6). Injection of 30 μL of prepared serum sample (113.3 pmol/L) produced a mean S/N ratio of 33.7:1, with a CV of 5.9% (N=10); accordingly, the LOQ and LOD of the ID-LC–MS/MS method were 33.3 and 10.0 pmol/L, respectively.
No obvious matrix effect was observed for the four different types of serum samples as the ion intensity showed an average increase of 0.03%. The average ion intensity differences for normal, hemolytic, lipemic, and icteric serum samples were -0.45%, 0.74%, -0.61%, and 0.45%, respectively.
Analytical recovery, imprecision, and uncertainty
The analytical recoveries for the ID-LC–MS/MS method are listed in Table 1. The mean analytical recoveries and CVs were 100.6%±0.3%, 100.6%±0.3%, and 99.8%±0.9% for 1.14, 1.83, and 3.43 ng added C-peptide, respectively.
The within-run, between-run, and total precision of the ID-LC–MS/MS method were in the ranges of 1.0%–2.1%, 0.6%–1.2%, and 1.3%–2.2%, respectively, at four levels (199.3-2,836.8 pmol/L) (Supplemental Data Table S2). The measurement uncertainty estimated at four levels is shown in detail in Supplemental Data Table S3. The relative measurement uncertainties ranged from 3.7% to 4.6% at C-peptide concentrations of 199.3–2,836.8 pmol/L.
Method comparison
The imprecision of the six routine immunoassays is presented in Supplemental Data Table S4. One outlier identified for the Beckman assay was excluded. OLR and BA plots of the agreement between the six routine immunoassays and the ID-LC–MS/MS method are shown in Fig. 2. All R2 values were >0.98, but most slopes deviated from 1.0, with intercepts ranging from -43.7 to -234.4 (Table 2). All assays, except the Siemens and Beckman assays, displayed significant positive bias according to the BA analysis results (49.9%, 35.4%, 32.2%, and 81.7% for Roche, Abbott, Mindray, and Snibe, respectively; Table 2).
Recalibration
After recalibration using five frozen serum pool samples assigned by the ID-LC–MS/MS method, the slopes in OLR analysis of the agreement between the developed ID-LC–MS/MS method and the six routine assays were much closer to 1.0 (1.03-1.09) and the intercepts much closer to 0 (Table 2, Supplemental Data Fig. S1A, S1C, S1E, S1G, S1I, and S1K). The mean relative difference between the ID-LC–MS/MS method and the routine assays was remarkably reduced (-6.0%–1.2%) (Supplemental Data Fig. S1B, S1D, S1F, S1H, S1J, and S1K).
DISCUSSION
To improve the harmonization status of serum C-peptide measurements in China, we, the Chinese NCCL, developed and validated a reliable ID-LC–MS/MS (pmol/L) method that can be used as an accuracy base for routine serum C-peptide measurements. Accurate ID-LC–MS/MS-based methods for serum C-peptide measurement require suitable sample preparation procedures, which should provide sufficient sensitivity and minimal matrix effects. Our sample preparation procedure (inspired by studies conducted by Stoyanov, et al. [16, 17]) enables simple and fast purification of C-peptides from serum while ensuring high recovery and specificity. We used ZnSO4 solution instead of an organic solvent to precipitate proteins, which allows the direct loading of the supernatant to the SPE cartridge and eliminates the need for additional procedures to remove the organic solvents before SPE separation.
Because of the low fragmentation efficiency of C-peptide in MS/MS, which can result in a significant reduction in sensitivity, optimizing sensitivity is crucial for the development of ID-LC–MS/MS-based methods for C-peptide measurement [18]. We found that adjusting the Q1 resolution from unit to low can enhance the signal of the product ion by nearly four-fold (data not shown). One possible explanation is that when the Q1 resolution is unit, only part of the isotopic distribution of the precursor is captured due to the narrow isolation window. After adjusting the Q1 resolution to low, a larger part of the precursor isotopic distribution is allowed to enter the collision cell, generating more abundant signals.
C-peptide is readily adsorbed to the surfaces of plastic and glass vials, and the strong absorption can be inhibited by adding non-specific binding proteins or sample matrix. We used FBS not containing human C-peptide to prepare the standard solutions and IS solutions, and the behaviors of the standard solutions were found to be similar to those of serum samples.
Desirable specifications of C-peptide measurement for routine assays are 8.3% for imprecision and 7.1% for inaccuracy, according to the Westgard website (https://www.westgard.com/biodatabase1.htm). Our ID-LC–MS/MS method obtained a desirable precision of ≤2.2% at four levels, which was less than 1/3rd of the imprecision criteria for routine methods. In the absence of international serum CRMs for C-peptide in the JCTLM database, we conducted a spiking recovery experiment to validate the accuracy of our method, and acceptable recoveries with outstanding CV% were observed (99.6%–101.0%; CV, 0.3%–0.9%), which was less than 1/4th of the accuracy criteria for routine assays. The excellent recovery demonstrated that our method recognizes the analyte in samples and provides stable measurement signals for the identified analyte. However, the trueness and accuracy of quantification also depend on the accuracy and traceability of the calibrator. The calibrator used in our method was prepared from the primary CRM for C-peptide in the JCTLM list, which has certified purities and uncertainties and can be traced to the SI unit. Given these characteristics, our method was substantially more accurate and precise than the routine assays and can be used as an accuracy base of routine assays.
Strong correlations were found between the six routine assays and the ID-LC–MS/MS method, with all R2 values >0.98. However, except for the Beckman and Siemens assays, the slopes for most assays deviated from 1.0 (1.54-1.88), which was consistent with the large relative differences (32.2%–81.7%). Among the six routine assays, the Beckman and Siemens assay showed better agreement with our ID-LC–MS/MS method than the other assays. One possible explanation is that the WHO materials are pure C-peptide solutions, and C-peptide may behave differently in a pure solution than in a sample-matrix solution, which may induce different matrix effects for different measurement methods. Both the Beckman and Siemens assays are magnetic particle chemiluminescence immunoassays. The WHO material may have a lower matrix effect for magnetic particle chemiluminescence immunoassays than for other assay methods. Another possible explanation is that the WHO material was a pure C-peptide powder, and the users had to prepare calibrator solutions themselves. Considering that C-peptide is unstable at room temperature (<24 hours) and readily adsorbs to vial surfaces, different manufacturers may induce different degrees of variation during calibrator preparation.
The consistency among the routine assays was effectively improved after recalibration with the five serum pool samples assigned by our ID-LC–MS/MS method, as the slopes were substantially closer to 1 (1.03-1.09) after recalibration and the percentage differences were greatly minimized. Therefore, the inconsistency among different immunoassays may mainly be due to the valuation transfer and traceability system. Therefore, the comparability among laboratories in China could be greatly improved by tracing their measurement results using our ID-LC–MS/MS method.
In conclusion, the Chinese NCCL developed a new, accurate, and precise ID-LC–MS/MS method to measure serum C-peptide concentrations. The method can be used to assign values to QC materials in the EQA program, and combined with joint endeavors of manufacturers and clinical laboratories, is expected to allow harmonization of clinical C-peptide measurements in China.
 XML Download
XML Download