

 PDF
PDF Citation
Citation Print
Print



Introduction
Biliary tract cancer (BTC) typically includes intrahepatic, perihilar, or extrahepatic cholangiocarcinoma (ECC) and gallbladder cancer (GBC) and shows poor 5-year survival rates of 15%–30%, depending on its primary location [1]. Surgical resection remains the only curative treatment option for early-stage disease, but the treatment options for advanced or metastatic BTC are limited [2].
As with other advanced or metastatic cancer types, systemic chemotherapy is a standard of treatment for advanced or metastatic BTC, but this has shown limited efficacy with low response rates in this disease, despite the advance in therapeutic strategies [3]. Therefore, a deep understanding of the biological characteristics and pathogenesis of BTC is essential to develop new therapeutic options, thereby improving the prognosis of patients with BTC. Employing appropriate in vitro and in vivo models is necessary to identify new treatment regimens, suitable cancer cell lines, or cultured cells; also, patient-derived xenograft (PDX) models established from human BTC may represent useful tools for BTC research, including prediction of efficacy and mechanisms of action of new therapeutic agents.
However, there are few BTC cell lines or cultures available for research, and the lack of these in vitro models has not sufficiently reflected the diversity of BTC phenotypes in experiments [4]. Moreover, establishing animal models of BTC is essential for validating data for experiments using in vitro models. The PDX mouse model has been considered a useful preclinical animal model in various tumors [5]. To date, there are a few PDX models engrafted by human BTC tissue, and there is an absolute need for establishing PDX models and in vitro models from various BTC cases [6].
Obtaining tumor tissue from primary or metastatic tumor sites in patients with BTC has not been regarded as readily achievable because these patients often undergo percutaneous needle or endoscopic biopsy for tumor tissue, leading to a low percentage of tumor cells in the sample. However, malignant ascites led by peritoneal invasion identified up to 10%–20% of cholangiocarcinoma patients at diagnosis, are easy to acquire and contains neoplastic cells, which makes malignant ascites useful tools alternative to tissue biopsy of metastatic tumors [7]. Recently, malignant ascites was reported to be a useful source of patient-derived tumor cell (PDC) in various cancers with a higher success rate [8]. Similarly, there are several reports regarding the successful establishment of malignant ascites-derived cancer cell lines or PDX models [9,10]. However, there have been few attempts to establish patient-derived cancer cell cultures or xenograft models from malignant ascites in patients with BTC.
This study established patient-derived cancer cell cultures and PDX models from malignant ascites in five patients with BTC and peritoneal spread. Given that commercially available BTC cell lines lack information about their genetic characteristics, which has made barriers to the development of targeted therapy, we characterized some of the preclinical models for genetic alterations using next-generation sequencing (NGS) analysis in this study.
Materials and Methods
1. Patients and sample collection
Between July 2017 and February 2018, five patients (AMCBTC-01, -02, -03, -04, and -05) with metastatic BTC were enrolled in this study. The inclusion criteria were as follows: age ≥ 18 years; histologically confirmed BTC; the presence of peritoneal spread leading to malignant ascites required to be drained by percutaneous approach. Approximately 500-mL ascites was collected at the time of paracentesis or other percutaneous drainage after all patients signed an informed consent form.
2. Isolation and culture of patient-derived BTC cells
About 500-mL ascites from each patient was centrifuged at 2,000 rpm for 5 minutes and the supernatant was removed. The pellet was washed using phosphate buffered saline (PBS) containing 2% fetal bovine serum (FBS), and the cells were counted. To establish primary cancer cell cultures, 5×105 cells were cultured on 60 mm collagen I-coated plates (Corning, Corning, NY) at 37°C in DMEM/F12 medium containing 2% FBS, 5 ng/mL epidermal growth factor (Invitrogen, Carlsbad, CA), 0.3 μg/mL hydrocortisone (Sigma-Aldrich, St. Louis, MO), 0.5 ng/mL cholera toxin (Sigma-Aldrich), 5 nM 3,3′,5-triiodo-L-thyronine (Sigma-Aldrich), 0.5 nM β-estra-diol (Sigma-Aldrich), 5 μM isoproterenol hydrochloride (Sigma-Aldrich), 50 nM ethanolamine (Sigma-Aldrich), 50 nM O-phosphorylethanolamine (Sigma-Aldrich), 1× insulin/transferrin/selenium (Invitrogen), and 1% penicillin/streptomycin until confluent, and sub-cultured at least twice before cryopreservation.
3. Establishing PDX mouse models
To derive a PDX mouse model for tumorigenicity assay, 3×106 cells in 100-μL PBS or matrigel were implanted into an inguinal mammary fat pad of 4–6-week-old immunodeficient female NOD.CB17-Prkdcscid (NOD-SCID, obtained from Koatech Inc., Seoul, Korea) mice. Body weight and tumor growth were monitored once a week. When tumors grew up to 1 cm in diameter, they were excised, and some fragments were cryopreserved for future analysis, while others were re-transplanted to NOD.CB17-Prkdcscid mice for the establishing stably transplantable xenograft. For future experimentation or expansion, engrafted tumors of the stably transplantable xenograft were cryopreserved in a freezing medium. Tumor tissues of PDX mice were stained for hematoxylin and eosin (H&E) staining.
4. DNA extraction for targeted NGS
The formalin-fixed, paraffin-embedded (FFPE) tissues for targeted NGS were reviewed by pathologists following matched H&E staining. According to our previous report, genomic DNA was extracted depending on the sample size and tumor cellularity [11]. Briefly, 2–5 sections (6 μm thick) from the indicated area in each FFPE tissue were obtained. After de-paraffinization with xylene and ethanol, gDNA was isolated using the NEXprep FFPE Tissue Kit (#NexK-9000, Geneslabs, Seongnam, Korea). Finally, quantification was performed using the Qubit dsDNA HS Assay kit (Thermo Fisher Scientific, Waltham, MA).
5. Targeted sequencing analysis
To evaluate somatic mutations, we used the NextSeq platform (Illumina Inc., San Diego, CA) with OncoPanel AMC v.3 described in our previous paper [12]. Two hundred nanograms of all gDNA, except for three BTC patient primary tissues (AMCBTC-01, -02, and -04) that used gDNA less than 30 ng, were fragmented using adaptive focused acoustic technology (Covaris Inc., Woburn, MA) to average fragment size distribution between 150 and 250 bp, followed by size selection using Agencourt AMPure XP beads (Beckman Coulter, High Wycombe, UK). Each DNA library was prepared by sequential reactions of end repair, A-base-tailing, and ligation with a TruSeq adaptor, using a SureSelectXT Reagent kit (Agilent Technologies, Santa Clara, CA). All libraries were addressed with sample-specific barcodes of 6 bp and quantified using a Qubit dsDNA HS Assay kit. Eight libraries were pooled to 750 ng for hybrid capture using an Agilent SureSelectXT custom kit (OP_AMCv3 RNA bait; Agilent Technologies). The concentration of the enriched target library was measured by quantitative polymerase chain reaction (PCR; Kapa Biosystems, Woburn, MA), and the sample was sequenced to 75 bp paired-end on the NextSeq platform using the NextSeq Mid-Output Kit v.2 reagent (Illumina).
6. Bioinformatics analysis
Sequence reads of mouse origin were identified and filtered out using Xenome v.1.0.0 [13]. Only read pairs with both reads classified as human were further analyzed in the same process as previously reported paper [12]. Mapping the sequenced reads was based on the human reference genome (National Center for Biotechnology Information build 37) with BWA (0.5.9) with default options. PCR duplicates were removed using MarkDuplicates of Broad Institute’s Picard package (available at https://broadinstitute.github.io/picard). Local realignment of deduplicated reads was performed at known indel positions using the GATK IndelRealigner tool. Next, base quality score recalibration was performed using the GATK BaseRecalibrator tool (https://software.broadinstitute.org/gatk/download). Somatic variant calling for variant candidates, including single nucleotide variants and short indels, were detected with matched patient primary tissue or patient-derived BTC cells using Mutect v.1.1.6 and the SomaticIndelocator tool in GAKT (Broad Institute). Common and germline variants from somatic variant candidates were filtered out with the common dbSNP build 141 (found in > 1% of samples), Exome Aggregation Consortium release 0.3.1 (http://exac.broadinstitute.org), and Korean Reference Genome database (http://152.99.75.168/KRGDB) and an in-house panel of normal variants. Final somatic variants were annotated using Variant Effect Predictor version 79 and converted to maf file format using vcf2maf (GitHub, https://github.com/mskcc/vcf2maf, last accessed February 14, 2018).
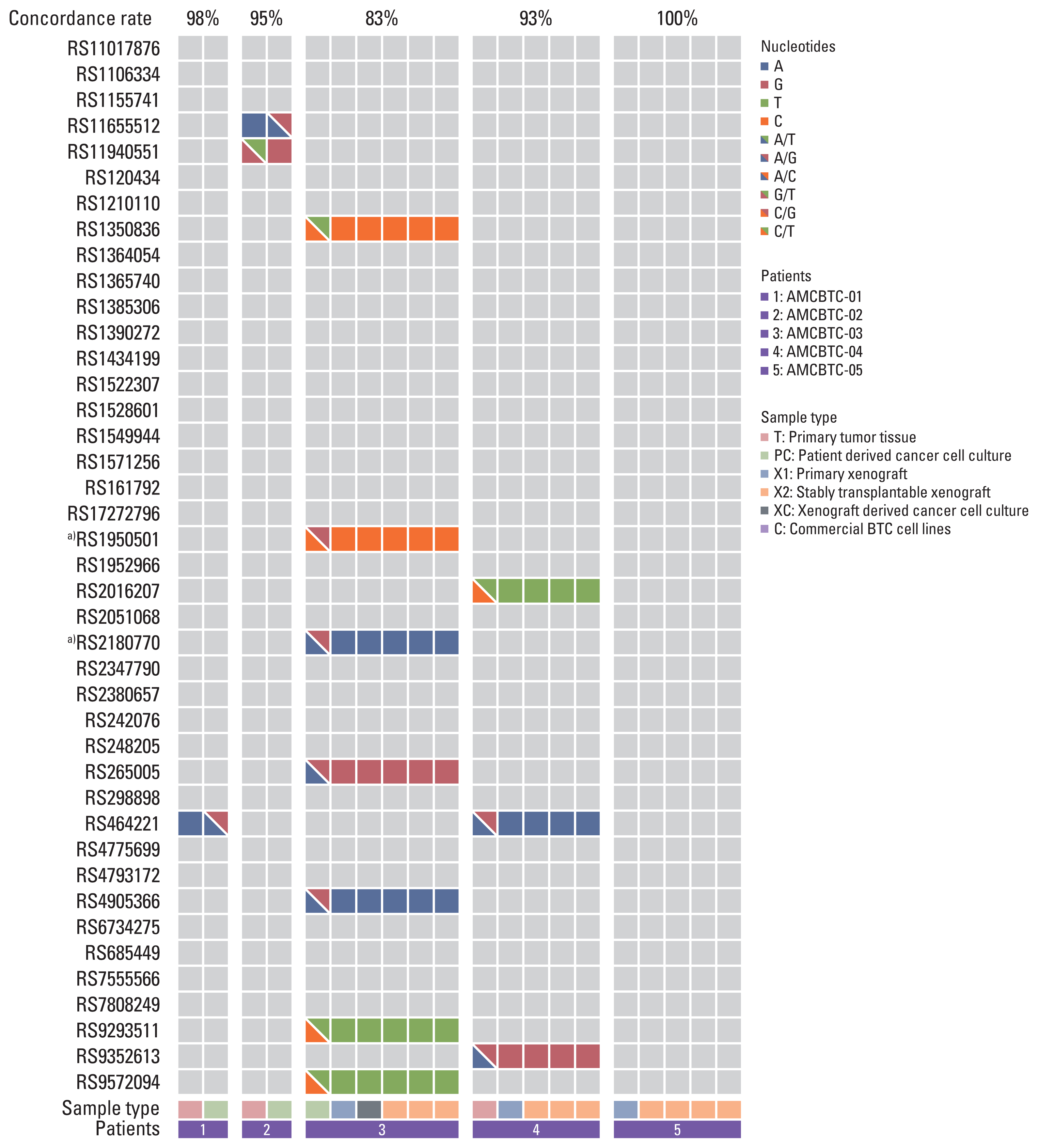
7. Single nucleotide polymorphism-based fingerprinting analysis
To validate whether patient-derived cancer cell cultures maintain the genetic characteristics of their original cancer tissue, the genetic fingerprinting comparison was performed between cancer cell cultures with matched primary tumor or PDX tumor tissues through genotyping for 48 known single nucleotide polymorphisms (SNPs) included in OncoPanel AMC v.1, which were used in our previous study [14]. The concordance between primary tumor or PDX tumor tissue and cancer cell culture in each patient (AMCBTC-01, -02, -03, and -04) was calculated. Samples with > 85% concordance were called a match.
8. Calculation of IC50 of gemcitabine and cisplatin in patient-derived cancer cell cultures from patients with BTC
The cells with growth medium were plated at a density of 2,000 cells in 50 μL per well of the 96-well plate. After 24-hour incubation, the drugs were treated into the cells and incubated for 72 hours. Cell viability was evaluated using the CellTiter-Glo assay (Promega, Madison, WI). Then, 20 μL of CellTiter-Glo assay solution was added into each well of the 96-well plate containing the cells in 100-μL culture medium and mixed for 2 minutes on an orbital shaker for cell lysis. The test plates were incubated with the plate at RT for 10 minutes to stabilize the luminescent signal, and then luminescence was read with PerkinElmer VICTOR X2, and IC50 values were calculated using GraphPad Prism 5.0 (GraphPad Software Inc., San Diego, CA).
Results
1. Clinicopathological characteristics of patient-derived cancer cell cultures and PDXs
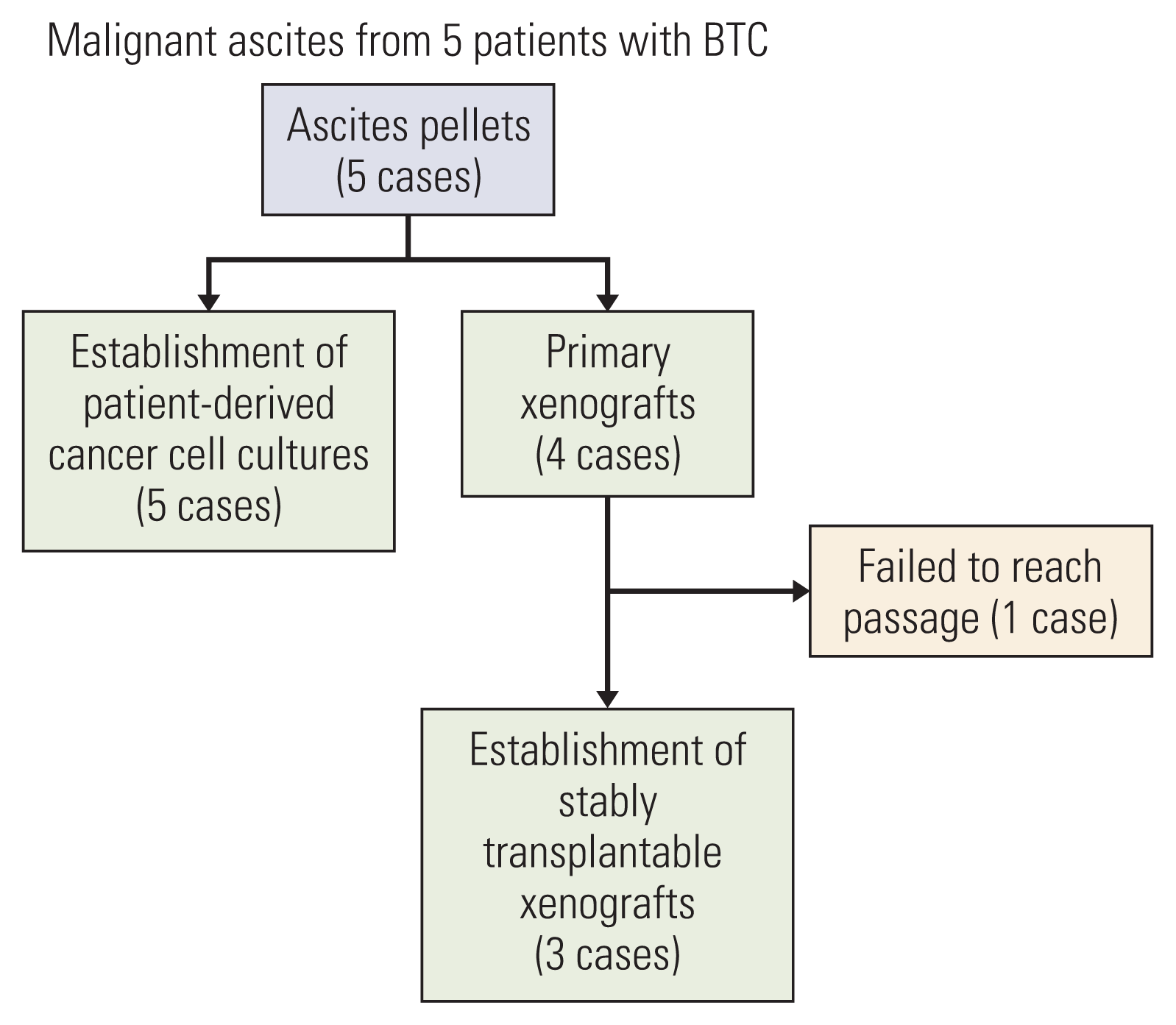
Malignant ascites were collected by paracentesis at the time of progression on second-line chemotherapy in all patients when clinically required. The workflow of patient-derived cancer cell cultures and PDXs from five patients with metastatic BTC is shown in Fig. 1.
Patient-derived cancer cell cultures were successfully established from ascites samples of five patients with BTC. For PDXs, four primary xenografts (from AMCBTC-02, -03, -04, and -05) were obtained by directly engrafting the viable tumor cells from malignant ascites fluid into mice. Subsequently, three xenografts (AMCBTC-03, -04, and -05) were successfully established as PDX models from re-transplantation of grown tumor tissue into new mice. The success rate of establishing patient-derived cancer cell cultures was 100% (five of five attempts). For PDXs, initial take rates (defined as initial outgrowth directly from patients) was 80% (four of five attempts), and success rates of generating stably transplantable xenografts (defined as PDX with the ability to be re-transplanted from initially transplanted mouse) was 75% (six of eight attempts).
Clinicopathological characteristics of five patients with histologically proven BTC are shown in Table 1. The cancer types were GBC (n=3, 60%) and ECC (n=2, 40%), and all patients had peritoneal spread as one of the metastatic sites. All five cases showed progressive disease to second-line chemotherapy (gemcitabine plus cisplatin followed by 5-fluorouracil/leucovorin containing regimen) at the time of ascites collection. The clinicopathological features of commercially available cell lines are summarized in Table 2.
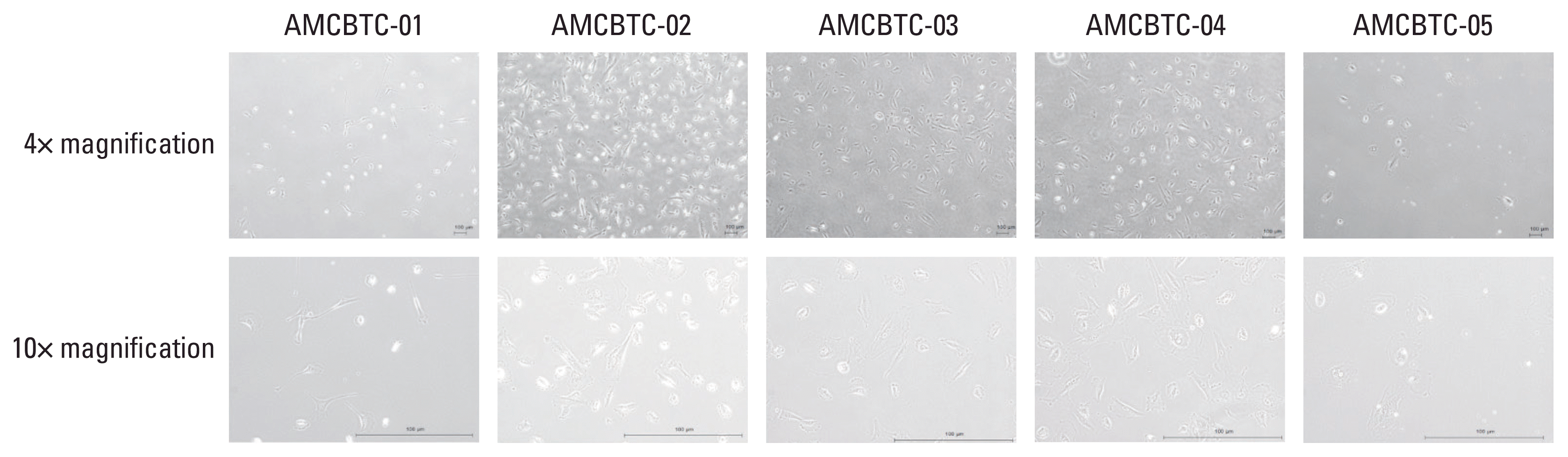
2. Cytological characteristics of patient-derived cancer cell cultures
The AMCBTC-01, -02, -03, -04, and -05 cell cultures grew as a monolayer of adherent cells (Table 3). Cytologically, AMCBTC-01 and -02 cell cultures grew primarily as spindle-shaped with long cytoplasm in vitro, but AMCBTC-03, -04, and -05 cell cultures grew in a pleomorphic shape (Table 3, Fig. 2). Additionally, the viability of these cells is described in S1 Table.
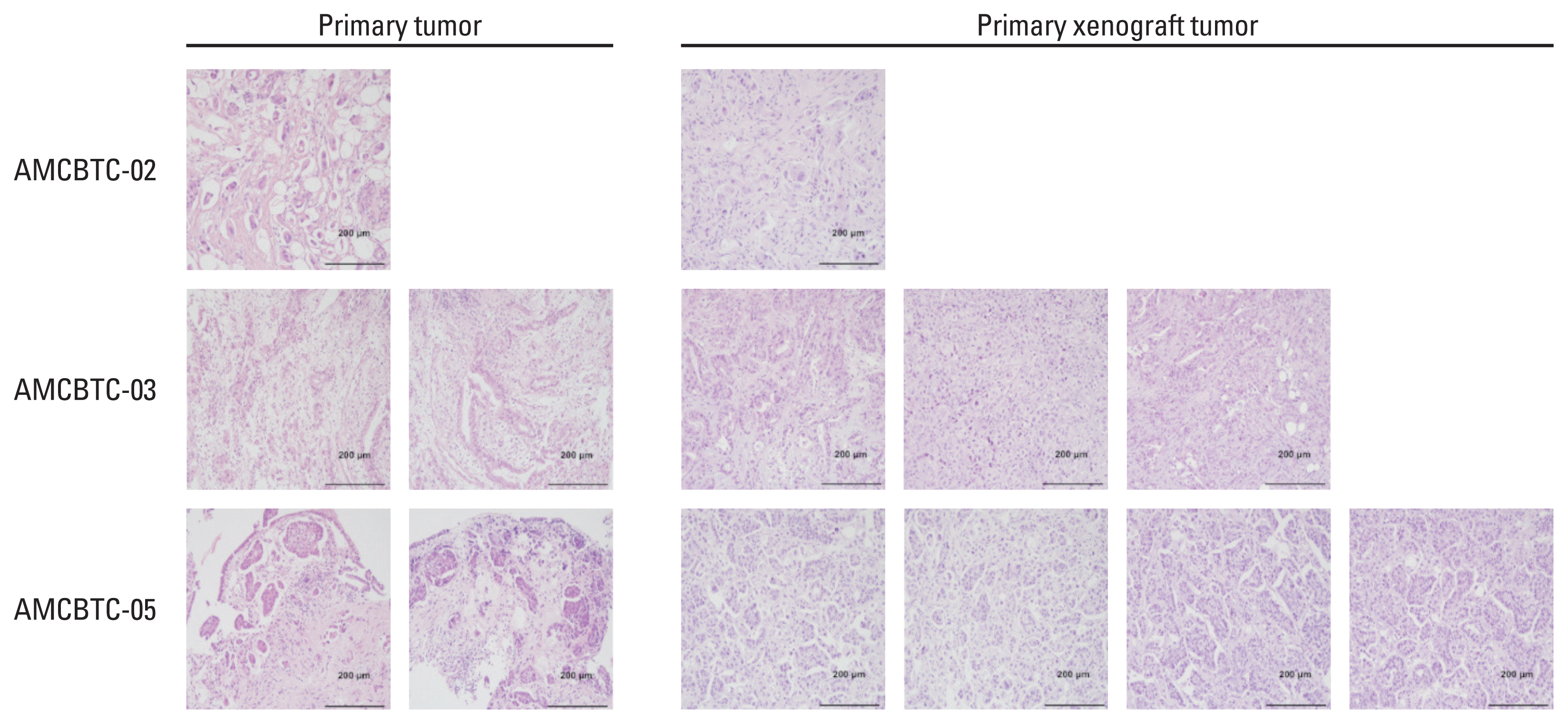
3. Comparing histopathology between primary tumor and primary xenograft tumor
We compared the histopathology between primary tumor and primary xenograft tumors to examine whether primary xenograft tumors remain the histological features of their parent tumors. Three pairs (AMCBTC-02, -03, and -05) were available for comparison among the four pairs in which primary xenografts were successfully obtained. However, the other pair was unavailable for comparison due to the lack of tumor components in the primary tumor specimen (AMCBTC-04). Comparing the morphological characteristics between primary tumors and primary xenograft tumors, we observed similar morphology in each pair (Fig. 3). In the AMCBTC-03 case, both primary and PDX tumor tissue shared the same morphology showing glandular formation, and there were characteristic clustered tumor cells without glandular formation in both tumor tissues of the AMCBTC-05 cases. These findings suggest that primary xenograft tumors are representative of matched primary tumors.
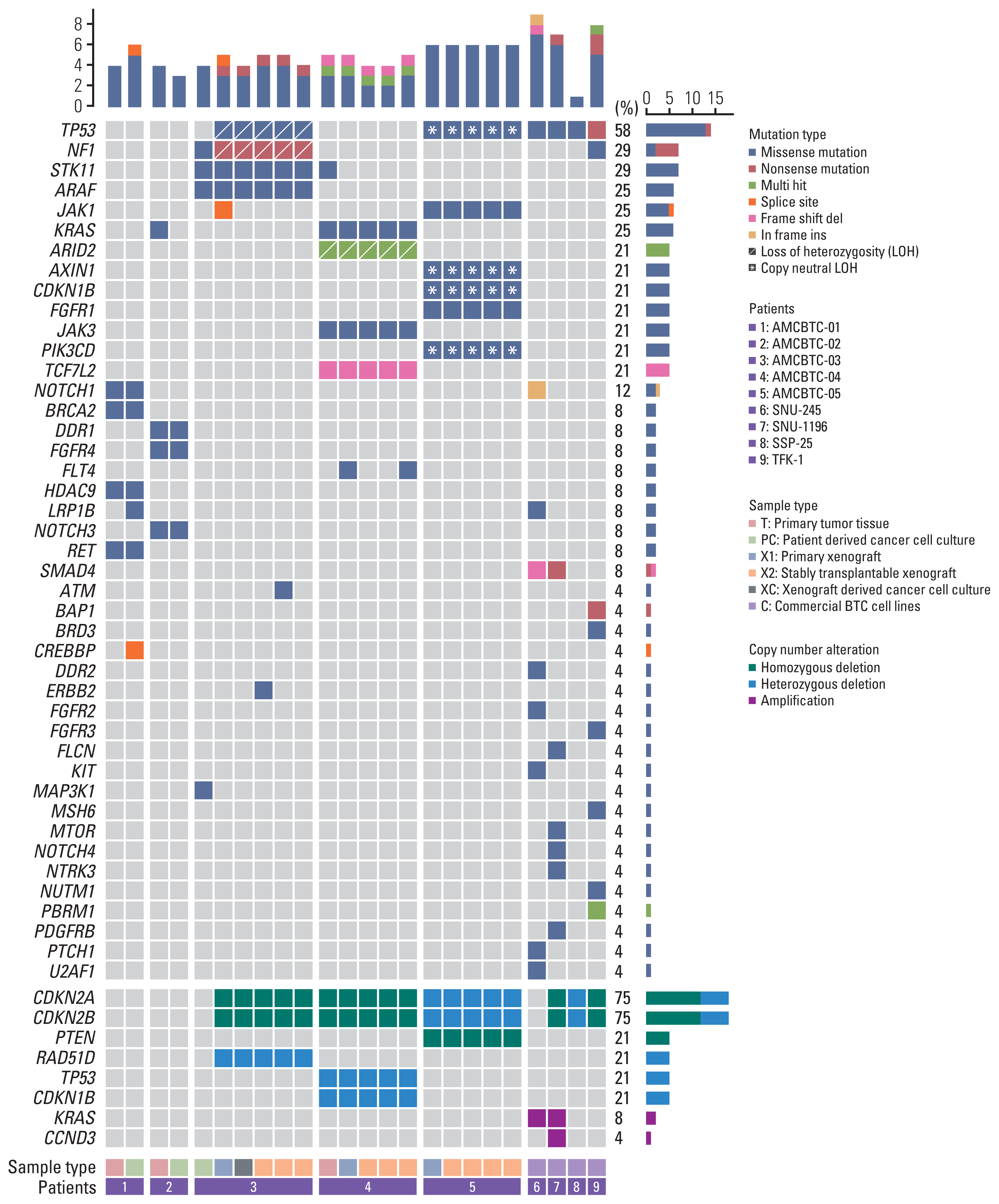
4. Genomic landscapes of the primary tumor, patient-derived cancer cell cultures, and PDX tumor
Genetic profiling was obtained in 20 samples (patient-derived cancer cell cultures, PDX models, or primary tumor tissue) from five patients with BTC available for targeted NGS analysis (primary tumor of AMCBTC-01, -02, and -04, patient-derived cancer cell cultures of AMCBTC-01, -02, and -03, and PDXs of AMCBTC-03, -04, and -05) (Fig. 4). Among the five primary tumor tissues and five patient-derived cancer cell cultures, two primary tumor tissues (AMCBTC-03 and -05) and two patient-derived cancer cell cultures (AMCBTC-04 and -05) failed during NGS analysis due to the lack of tumor component in primary tumor specimen and low cellularity of cultured cancer cells, respectively. Additionally, out of the four primary xenografts by directly engrafting human tumor cells into mice, one xenograft (AMCBTC-02) was unavailable for NGS analysis due to a lack of tumor component in tumor tissue.
We identified 51 genetic alterations in 48 genes from primary tumor tissues and patient-derived cancer cell cultures and PDX models of BTC patients; most of these alterations were nonsynonymous substitutions. The mutated genes observed in the samples (primary patient-derived cancer cell cultures and PDXs or primary tumor tissues) from more than one patient were TP53 (n=2), KRAS (n=2), JAK1 (n=2), and STK11 (n=2). Other mutated genes found in the samples from one of five patients with BTC were ATM, ARAF, AXIN1, ARID2, BRCA2, CDKN1B, CREBBP, DDR1, ERBB2, FGFR1, FGFR4, FLT1, FLT4, HDAC9, JAK3, LRP1B, MAP3K1, NF1, NOTCH1, NOTCH3, PIK3CD, RET, and TCF7L2. Concerning copy number alterations, CDKN2A and CDKN2B deletions were identified in all PDX tumor tissues from three patients (AMCBTC-03, -04, and -05), and RAD51D, CDKN1B, and PTEN deletions were observed only in tumor tissues of PDX from AMCBTC-03, -04, and -05, respectively. While most genetic alterations of primary tumor tissue remained in their patient-derived cancer cell cultures or PDX tumor tissues, there were differences in genetic alteration between patient-derived cancer cell culture and tumor tissues of corresponding PDX derived from one patient (AMCBTC-03). Unlike the mutation profile of patient-derived cancer cell culture of AMCBTC-03, those of tumor tissues from the PDX showed TP53 mutation and a nonsense mutation in NF1 with 100% variant allele frequency, suggesting a loss of heterozygosity (LOH) in these genes.
5. Comparing frequently altered genes between patient-derived cancer cell cultures and commercially available BTC cell lines
In this study, the overall pattern of commonly altered genes in primary patient-derived cancer cell cultures from three BTC patients was different from that in four commercially available BTC cell lines. For example, the SMAD4 and TP53, known as the frequently mutated genes in BTC, were mutated in half of and all commercially available BTC cell lines, respectively; however, no primary patient-derived cancer cell cultures showed these mutations. In contrast, several mutated genes (NOTCH3, STK11, ARAF, RET, DDR1, FGFR4, HDAC9, CREBBP, and BRCA2) only exist in primary patient-derived cancer cell cultures (AMCBTC-01, -02, and -03).
6. Patient-derived cancer cell cultures and PDX authentication by SNP fingerprinting analysis
The pattern of genomic alterations between patient-derived cancer cell cultures or PDXs and their matched primary tumor specimen was retained (Fig. 4). Furthermore, this result agrees with the result of SNP fingerprinting analysis (Fig. 5), showing that SNPs of primary patient-derived cancer cell cultures (AMCBTC-01 and -02) and PDX (AMCBTC-04) were mostly matched with those of its original primary tumor. This suggests that primary patient-derived cancer cell cultures and PDXs were derived from the same clone with its primary tumor tissues. However, seven SNPs of primary patient-derived cancer cell culture were not matched with those of paired PDX in AMCBTC-03, meaning that these samples have a lower probability of having the same clone. Moreover, these unmatched SNPs were from transitional changes in PDXs, leading to LOH in specific genes in PDXs, such as RAD51B and RIPK3. The primary tumor tissue was not enough for this analysis; thus, we only compared SNPs of patient-derived cancer cell culture and those of PDX tumors from the patient (AMCBTC-03).
7. Gemcitabine and cisplatin sensitivity evaluation using primary patient-derived cancer cell cultures
The IC50 value for all five primary patient-derived cancer cell cultures (AMCBTC-01, -02, -03, -04, and -05) was calculated, and the results are summarized in Table 4. Based on the IC50 values described in previous studies [4,15], primary patient-derived cancer cell cultures (AMCBTC-01, -02, -03, -04, and -05) seemed resistant to gemcitabine and cisplatin.
Discussion
BTC is heterogeneously malignant, but there are a limited number of BTC cell lines currently available with a lack of corresponding information and their unsatisfactory characteristics since the success rate for establishing BTC cell lines is low, usually about 10% [4]. Therefore, newly validated preclinical models are needed to improve our understanding of diverse molecular and genomic mechanisms of action in developing BTC cancer. This analysis reports on successfully established five primary patient-derived cancer cell cultures and three stably transplantable PDXs derived from malignant ascites in five patients with metastatic BTC. Each primary patient-derived cancer cell culture shows uncommon and different patterns from previous commercially available BTC cell lines in its tumor mutation profile. However, the genomic alterations and histologic features of PDX tumor specimens were similar to those of the corresponding parental tumors, suggesting that these preclinical models reflect the primary tumors in patients with metastatic BTC.
In this study, we attempted to demonstrate that malignant ascites fluid can be an effective source for establishing in vitro models, such as primary patient-derived cancer cell lines and PDX models with the preservation of histopathological features and genetic profiles of their parental tumors. Processing ascites with a culture medium is a common method for establishing a cancer cell line, and malignant ascites were reported to be a useful source of PDC or cancer cell lines in various types of cancers [8,10]. Additionally, several reports have established cancer cell lines from malignant ascites in BTC [16,17]. However, those cell lines were established more than 20 years ago, and there was limited information about their molecular and genetic characteristics. Therefore, this study presents novel PDX models and primary patient-derived BTC cell cultures and evaluates their genetic characteristics compared with previous studies.
Notably, according to SNP fingerprinting analysis, there was an unmatched result in seven SNPs of samples from one patient (AMCBTC-03). These mismatched SNPs of primary patient-derived cancer cell culture and tumor tissue of its corresponding PDX model from AMCBTC-03 were heterozygous and homozygous, respectively. As we removed all mouse reads identified using the mouse reference genome when analyzing the genetic mutation profile of PDX tumor tissues, there is a possibility that LOH occurred during engraftment in PDX models rather than contamination of mouse nucleic acid by mouse stroma. Interestingly, two genes (RAD51B and RIPK3) had mismatched SNPs, and these genes showed LOH. RAD51B is involved in the homologous recombination repair pathway of double-stranded DNA breaks by encoding the protein essential for DNA repair [18]. Furthermore, RIPK3 encodes a serine/threonine kinase and plays a crucial role in necroptosis, and it promotes antitumor immunity; there have been several reports suggesting that RIP3K expression is inhibited in various cancers and cancer cell lines [19]. However, as they were intronic mutations, it is challenging to determine the potential for pathogenicity of mutations in these genes with these SNP fingerprinting results. Nonetheless, there seem to be more LOH events in PDX models than those in their corresponding BTC cell cultures or primary tumor tissue in this study. This finding agrees with previous reports with PDXs derived from BTC, which showed that LOH was common in 26 PDXs available for genomic analysis among 47 successfully engrafted PDXs mostly derived from resected tumors [20].
Interestingly, there were TP53 missense mutations with LOH in each PDX model from two patients (AMCBTC-03 and -05), while there was no case of TP53 missense mutation in the primary tumor tissue or primary patient-derived cancer cell cultures available for NGS analyses (Fig. 4, S2 Table). The TP53 is a tumor suppressor gene, and mutation in this gene is frequently found in human BTC with a range of 26% to 45%, depending on the anatomical site of BTC [21]. Although the absence of NGS analysis for their corresponding primary tumor tissue due to the lack of tumor components in these patients does not confirm whether this missense mutation with LOH was newly developed, the other alterations in genes related to tumor suppressor or apoptosis (NF1, AXIN1 mutation with LOH, CDKN2A, and CDKN2B deletion) only existing in the PDX models suggest that LOH of TP53 might be newly developed during PDX engraftment. Furthermore, TP53 LOH has been reported to be a critical prerequisite for stabilization and gain-of-functions of mutant p53 protein in mouse tumors carrying missense TP53 mutation and in several sporadic human cancers using public human genomic databases (TCGA and METABRIC) [22]. Altogether, these results suggest that the occurrence of LOH involving the particular genetic loci related to tumor suppressor or apoptosis in PDX tumor tissues was likely to happen during PDX establishment as a way to provide a favorable environment for cancer growth.
This study compared the frequency and pattern of altered genes in patient-derived cancer cell cultures with those in commercially available BTC cell lines. Furthermore, we found that the main genetic alterations of several known oncogenes and tumor suppressor genes, including KRAS, TP53, and SMAD4, in commercially available BTC cell lines in this study were consistent with those in BTC cell lines (SNU-245, SNU-1196, and TFK-1) in the previous study [23]. Furthermore, compared with commercially available BTC cell lines, our preclinical models from five patients with BTC show a different pattern of genetic mutation profiles, conferring diverse characteristics of in vitro models for the study of BTC biology in terms of heterogeneity. Unlike the data of commercially available BTC cell lines, there were several mutated genes (NOTCH3, STK11, ARAF, RET, DDR1, FGFR4, HDAC9, and BRCA2) only existing in primary patient-derived cancer cell cultures (AMCBTC-01, -02, -03). These genetic mutations were reported to be relatively infrequent in molecularly characterized 260 cases of BTC reported by Nakamura et al. [21], and few molecularly-targeted therapeutics have been reported to be effective to tumors harboring these mutations [24]. However, among those mutations, a mutation in BRCA2 was identified in primary tumor tissue samples, and its corresponding patient-derived cancer cell culture was available for target-sequencing analysis from one patient (AMCBTC-01). The BRCA2 gene is involved in DNA damage repair mediated by homologous recombination with BRCA1 [25]. As BRCA 1/2 mutated cells accumulate double-strand DNA breaks and result in carcinogenesis through genomic instability, patients with germline mutations in BRCA 1/2 had about five times relative risk of developing BTC [25]. Additionally, BTC patients with mutations in homologous recombination repair-related genes, including BRCA 1/2 have been suggested to have increased sensitivity to DNA damaging therapeutic agents [26]. Therefore, preclinical models showing BRCA2 mutation might be a useful tool in this class of drug sensitivity tests.
In our study, the success rate of stably transplantable PDX using tumor cells from malignant ascites of BTC patients seems higher than those reported using primary tumor tissue (ascites vs. primary tumor tissue, 60% vs. 5.8%) [4,6,27]. Additionally, in this study, the success rate of patient-derived cancer cell culture was 100%, and this was higher than that of BTC cell line using primary tumor tissue (20%–45%) [27]. This agrees with the previous study establishing PDCs from human body fluids, such as ascites and pleural effusions, and the success rate of PDCs was more than 70% in the study [8]. Although the precise mechanisms for this higher success rate of ascites-derived cancer cell cultures and PDX models remain unclear, different tumor microenvironments (TMEs) between primary tumor tissues and ascites-derived tumor cells might be associated with this result. Tumor tissue consists of cancer cells and noncancerous components [28]. Notably, of the noncancerous components, fibroblast was reported to influence cancer cell proliferation positively or negatively depending on the fibroblast cell type and cell-to-cell interactions in TME [29]. Thus, a lack of TME consisting of fibroblasts and other noncancerous components in processed ascites-derived tumor cells might aid cancer cells to proliferate better positively. Notably, we transplanted tumor cells into the mammary fat pad of mice instead of subcutaneous transplantation in this study. The mammary fat pad has been used as an orthotopic transplantation site for the breast cancer PDX model, and evidence from a recent study has demonstrated that orthotopic implantation of the patient tumors into the mammary fat pad showed better engraftment and faster growth than subcutaneous implantation in breast cancer PDXs [30]. Because there have been few xenograft studies using mammary fat pad implantation in BTCs, it is hard to draw a firm conclusion that mammary fat pad implantation has a positive effect on tumor cell engraftment compared with subcutaneous implantation, leading to better success rates.
This study evaluated chemosensitivity tests for gemcitabine and cisplatin using primary patient-derived cancer cell cultures from five patients with BTC, and all of these were gemcitabine- and cisplatin-resistant. As all patient-derived cancer cell cultures in this study were derived from chemotherapy-resistant patients (gemcitabine and cisplatin), these susceptibility results to anticancer therapies show a close correlation with clinical characteristics of patients from which the patient-derived cancer cell cultures originated.
Our study has some limitations; first, some of the primary tumor tissues were not available for NGS analysis due to the lack of tumor portion in the specimen, thereby comparing the genetic profiles between primary tumor tissues, patient-derived cancer cell cultures, and PDX tumor tissues in each patient was impossible. Second, there are no established patient-derived cancer cell cultures or PDX models from patients with intrahepatic cholangiocarcinoma (ICC) classified as BTC subgroup. Therefore, we do not know whether cancer cell cultures or PDX from ICC could be established from malignant ascites with a higher success rate and maintain the histopathological structures and genetic characteristics of parental tumors. Third, implanting tumor cells into mammary fat pads to establish the xenograft model in this study is not typically used but an experimental method in establishing PDX models of BTC. As orthotopic implanting into mammary fat pads has been used in establishing the breast cancer PDX model, the current PDX models may not recapitulate the TME of BTC. And the characteristics of the PDX models using only female mice in this study need to be further evaluated in future studies with male mice. Lastly, information about genetic characteristics is based on a small number of models in a few patients with BTC, therefore this study may not be reflective of the characteristics in entire populations of subgroups in BTC.
Conclusively, we have now established five patient-derived cancer cell cultures and three stably transplantable PDX models from malignant ascites with molecular and clinical characterization from five patients with metastatic BTC. As an alternative to the primary tumor tissue for the source of preclinical models in BTC, malignant ascites are useful sources with a higher success rate. Furthermore, these models accompanied by different genetic characteristics from conventional BTC cell lines will be a useful tool for better understanding BTC biology. The preclinical models established in this study will be shared to the scientific community according to the relevant procedural guidance.
 XML Download
XML Download