

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Novel therapies targeting androgen receptor (AR) signaling are needed for metastatic castration-resistant prostate cancer (mCRPC). Poly (ADP-ribose) polymerase (PARP) inhibitors belong to a class of targeted agents under development for the treatment of homologous recombination repair (HRR)-deficient tumors [1]. PARP inhibitors block DNA damage repair by trapping PARP bound to DNA single-strand breaks, leading to replication fork stalling, causing collapse and generation of DNA double-strand breaks, ultimately resulting in cancer cell death [2]. The clinical efficacy and safety of a PARP inhibitor compared with those of an AR signaling inhibitor are being tested in patients with HRR-deficient mCRPC in a pre-chemotherapy setting in the ongoing phase III PROfound trial (NCT02975934) [3].
Somatic and/or germline assays for HRR gene mutations are performed with multigene panels to select patients with prostate cancer who are candidates for PARP inhibitor treatment. The incidence of pathogenic somatic and germline mutations in HRR genes differs between localized and metastatic prostate cancer [4]. Pritchard, et al. [5] identified germline mutations in 11.8% of DNA damage repair (DDR) genes among 692 patients with metastatic prostate cancer. This incidence was significantly higher than that among men with localized prostate cancer (4.6%) [6]. The second “hit” somatic aberration within the tumor genome was identified in 59% of patients with metastatic prostate cancer harboring germline DDR gene mutations. DNA repair pathway mutations are commonly detected in metastatic prostate cancer, with the prevalence of deleterious mutations in HRR genes reaching 28% [7, 8].
In May 2020, based on positive data from the phase III PROfound trial, olaparib was approved by the US Food and Drug Administration (FDA) for adult patients with deleterious or suspected deleterious germline or somatic HRR gene-mutated mCRPC who have progressive disease following prior treatment with enzalutamide or abiraterone. For the selection of patients with mCRPC carrying HRR genetic variants, the FDA also approved the FoundationOne CDx (Foundation Medicine, Inc., Cambridge, MA, USA) next-generation sequencing (NGS) panel, containing a prespecified set of 14 genes involved in the HRR pathway, which was used in the PROfound trial. In Korea, olaparib is also considered a potential treatment for HRR-deficient mCRPC. A validated NGS assay is needed to obtain reliable results to appropriately select candidates for PARP inhibitor treatment.
The multigene NGS panel for detecting HRR mutations has not been approved as a companion diagnostic tool in Korea. Validation of multigene NGS panels for the HRR pathway has not been performed in tissue materials of Korean patients with prostate cancer. The aim of this study was to validate locally adopted NGS assays (local laboratory) using samples from the PROfound trial for which genetic variants of HRR genes were previously evaluated using the FDA-approved FoundationOne CDx NGS assay (central laboratory) as the best available method [3]. By demonstrating comparable analytical performance of the local laboratory NGS assays, other NGS assays could be considered as options for HRR gene mutation analysis in patients with mCRPC.
MATERIALS AND METHODS
Study design
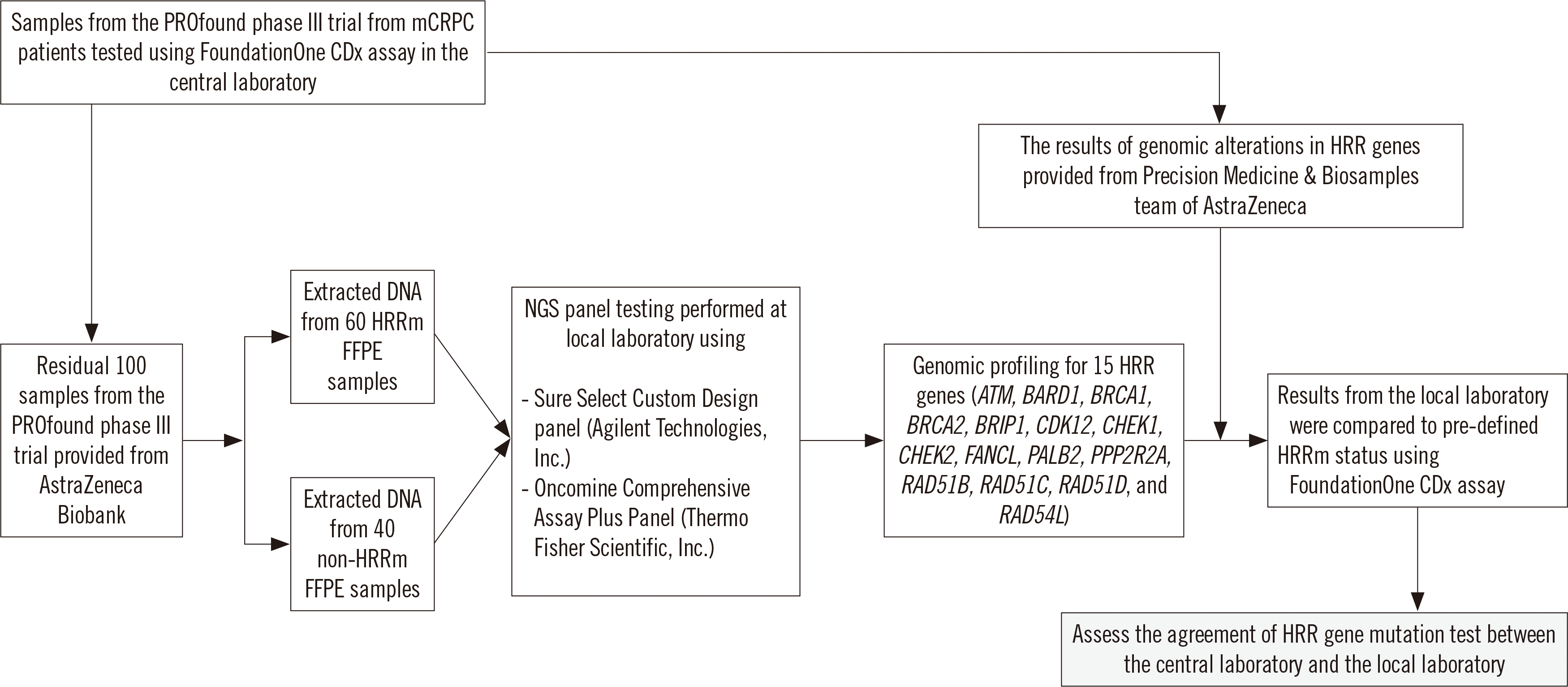
For genomic profiling of HRR-related genes, NGS assays were performed with the SureSelect Custom Design panel (Agilent Technologies, Inc., Santa Clara, CA, USA) and Oncomine Comprehensive Assay Plus Panel (Thermo Fisher Scientific, Inc., Waltham, MA, USA) on samples from the PROfound phase III trial. The generated genomic profiles of 15 genes (ATM, BARD1, BRCA1, BRCA2, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, and RAD54L) were compared with the genomic variant data of HRR genes discovered by the FoundationOne CDx assay in the PROfound trial (Fig. 1). This retrospective study, conducted from September 2020 to May 2021, was approved by the Institutional Review Board of Gangnam Severance Hospital, Seoul, Korea (3-2020-0326).
Clinical samples
A total of 100 samples from patients with mCRPC in the PROfound trial collected from January to December 2015 with patient consent were selected, including 60 HRR mutation (HRRm) cases, with deleterious variants on HRR pathway-related genes, and 40 non-HRRm cases with no such variants. The extracted genomic DNA of 100 PROfound formalin-fixed, paraffin-embedded (FFPE) tissue samples was provided by AstraZeneca Biobank, and shipped samples were stored at −80°C. The quality and concentration of DNA were assessed using a 2200 TapeStation instrument (Agilent Technologies) with the Agilent Genomic DNA Screen System and using a Qubit 3.0 Fluorometer with the QubitdsDNA HS Assay Kit (Thermo Fisher Scientific), respectively.
NGS and bioinformatics analysis
For NGS, a library was prepared using the Oncomine Comprehensive Plus assay (Thermo Fisher Scientific) targeting 500 oncogenes, including driver genes and tumor suppressor genes. This NGS assay is designed to detect single nucleotide variants (SNVs) and copy number variations (CNVs) in DNA and gene fusions in RNA. The libraries were prepared using nucleic acid input according to the Oncomine Comprehensive Plus user guide. The constructed library was used for templating and sequencing with the Ion 550 Kit on Ion Chef and the Ion S5 XL system (Thermo Fisher Scientific). Alignment to the hg19 human reference genome and variant calling were performed using Torrent Suite version 5.12.1 and Ion Reporter software version 5.18. The Torrent Suite software provides coverage analysis data, and Ion Reporter provides a report of annotated variants using Oncomine Comprehensive Plus–w2.3–DNA–Single Sample Workflow. The threshold was set to a minor allele frequency (MAF) ≥1.5%.
CNVs were analyzed on samples with a median absolute value of all pairwise differences (MAPD) of 0.5, which is a measure of read coverage noise detected across all amplicons in a sample. CNVs were called when the copy number ratio was <0.85 and P<10−5 representing copy number loss; these CNVs of HRR genes were visually reviewed with the copy number plot generated by Ion Reporter.
The SureSelect Custom Design panel assay was designed for detecting SNVs and CNVs in DNA, targeting 15 genes related to the HRR pathway. Genomic DNA samples were fragmented using Agilent’s SureSelect Enzymatic Fragmentation Kit (Agilent Technologies, Inc.), and capture probes were hybridized to target regions using SureSelect XT HS2 Target Enrichment Kit (Agilent Technologies, Inc.). The final SureSelect XT HS2 target-enriched libraries were sequenced using an Illumina NextSeq 550Dx platform (Illumina, San Diego, CA, USA) with the 2×150-bp paired-end read module. Sequencing data from the NextSeq 550Dx system were aligned to the hg19 human reference genome. The demultiplexed FASTQ data obtained using Illumina’s bcl2fastq software (https://support.illumina.com/downloads/bcl2fastq-conversion-software-v2-20.html) were further processed for alignment and annotation using a customized analysis pipeline. The threshold was MAF ≥1.5%.
The FoundationOne CDx assay was performed at Foundation Medicine, Inc. sites located in Cambridge, MA and Morrisville, NC (USA). Using the Illumina HiSeq 4000 platform, hybrid capture-selected libraries were sequenced to a high uniform depth. Sequence data were processed using a customized analysis pipeline designed to detect all classes of genomic variants. The threshold was MAF ≥5% (SNVs: MAF ≥1% at hotspots, indels: ≥3% at hotspots). The results of genomic variants in HRR genes were provided by the Precision Medicine and Biosamples team of AstraZeneca.
Candidate variants for comparison between platforms were selected only when the variant allele frequency (VAF) at a given position was ≥5% (SNVs: MAF ≥1.5% at hotspots or HRR mutation, indels: ≥3% at hotspots or HRR mutation). The allele frequency (%) was calculated by dividing the mutant coverage depth by the total coverage depth. For comparing CNVs, samples were analyzed by the R package ExomeDepth, which has been used to identify CNVs for germline and tumor samples [9, 10], available at the Comprehensive R Archive Network (CRAN) (https://cran.r-project.org/web/packages/ExomeDepth/index.html/). All deletions detected with a read.ratio of <0.8 were visually inspected using the ExomeDepth CNV plot tool.
Statistical analysis
High-quality variants reported from the central laboratory (Foundation Medicine, Inc.) were defined as true-positive variant calls for evaluating analytical performance. The overlapping regions of interest among the FoundationOne CDx assay, SureSelect Custom Design panel, and Oncomine Comprehensive Assay Plus Panel for the above-mentioned 15 genes were used to define true-negative variant calls.
Statistical analysis was performed using Microsoft Excel 2013 with the add-in program Analyse-it v5.01 (Microsoft Corporation, Redmond, WA, USA) and MedCalc software (https://www.medcalc.org/). Overall percent agreement (OPA), negative percent agreement (NPA), and positive percent agreement (PPA) were calculated as described in the CLSI guidelines EP12-A2 [11]. Results of all statistical analyses are presented using the 95% confidence interval (CI) and a two-sided P-value; P<0.05 was considered statistically significant.
RESULTS
Quality and concentration of DNA
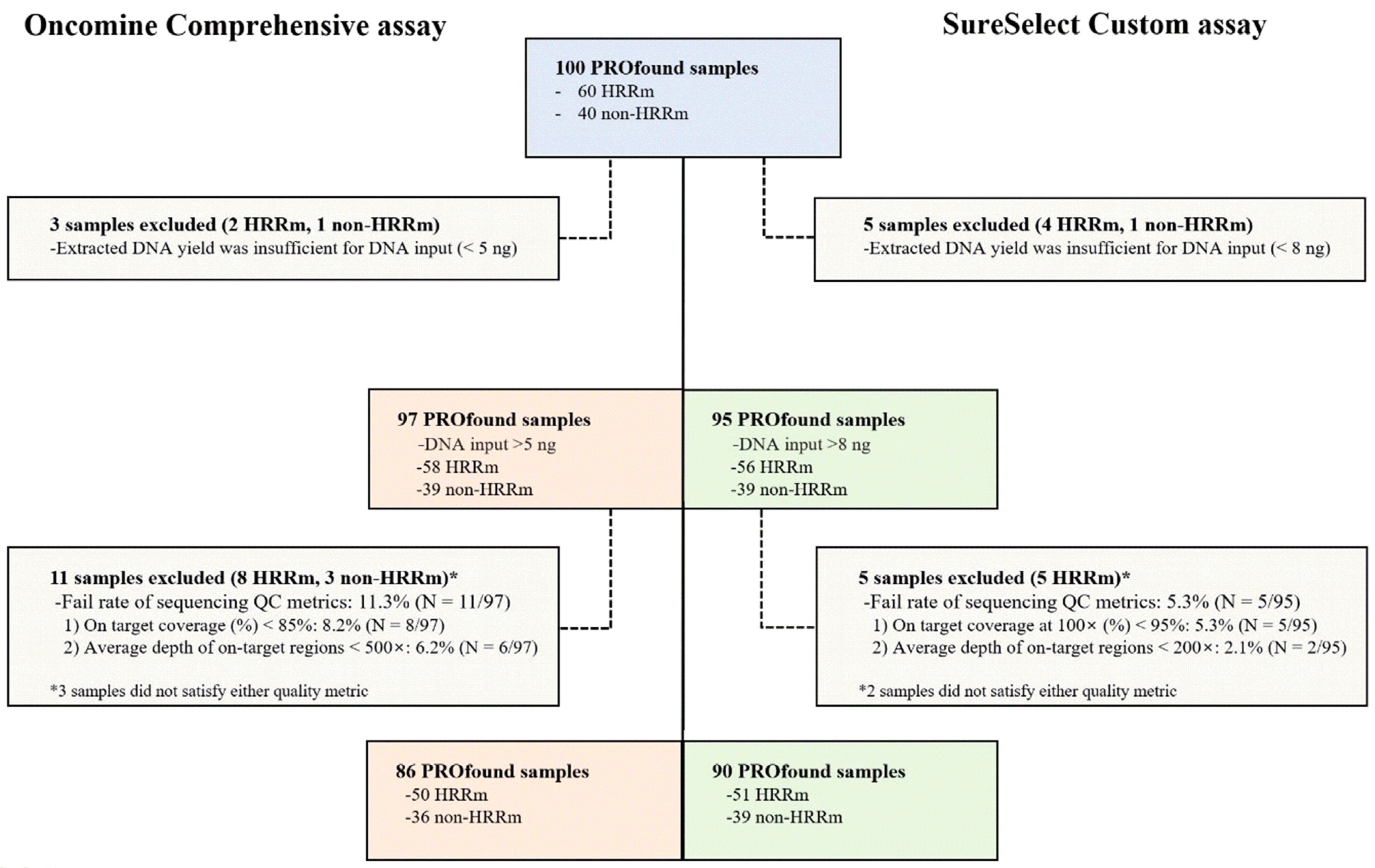
The DNA concentrations of samples are presented in Supplemental Data Fig. S1. The median DNA concentration measured using the Qubit 3.0 Fluorometer and a 2200 TapeStation Instrument were 4.4 ng/µL and 3.8 ng/µL, respectively, representing a significant difference (t-test, P<0.001). We determined the DNA inputs for an NGS assay based on the DNA concentration from the Qubit 3.0 Fluorometer according to the manufacturer’s instructions. Among the 100 PROfound samples, we used 97 samples for the Oncomine Comprehensive assay, and three samples with low DNA concentration (5 ng,<0.9 ng/µL) were excluded. We performed the SureSelect Custom assay using 95 samples (total DNA, >8 ng) (Fig. 2).
The average depth of on-target regions (> 500×) and on-target coverage (%) were used to assess the quality of sequencing for the Oncomine Comprehensive assay. Among the 97 PROfound samples, 86 (88.7%) satisfied the predefined sequencing QC metrics (Fig. 2) of the Ion S5 XL system. The average on-target reads and uniformity were 93.7% and 94.8%, respectively, in the Ion S5 XL system. The mapped reads and mean depth were 37,158,419 and 2,800×, respectively. The average on-target coverage at 100× and target bases with no strand bias reached 98.6% and 96.6%, respectively. In the SureSelect Custom assay, the average depth of on-target regions (>200×) and on-target coverage at 100× (%) were used to determine the quality of sequencing. Among 95 PROfound samples, 90 (94.7%) satisfied the predefined sequencing QC metrics (Fig. 2). The mapped reads and mean depth were 2,050,309 and 594×, respectively. The average on-target coverage at 100× reached 99.0%.
Comparison of the Oncomine Comprehensive and SureSelect Custom assays with the FoundationOne CDx assay
Three of the 100 samples with insufficient input DNA (<5 ng, 0.9 ng/µL) for the NGS assay and 11 of the remaining 97 samples that did not satisfy sequencing quality metrics were excluded from the performance evaluation of the Oncomine Comprehensive assay (Fig. 2). Finally, 86 PROfound samples (50 HRRm, 36 non-HRRm) were used for comparison of HRR mutation status between the FoundationOne CDx and Oncomine Comprehensive assays. The PPA, NPA, and OPA for SNVs and indels were 98.7%, 100%, and 100%, respectively (Table 1).
Among the 100 PROfound samples, 10 with insufficient input DNA and unsatisfactory sequencing quality metrics were excluded, and the 90 remaining PROfound samples (51 HRRm, 39 non-HRRm) were used for comparison of HRR mutation status between the FoundationOne CDx and SureSelect Custom assays (Fig. 2). The PPA, NPA, and OPA for SNVs and indels were all 100% between these two NGS assays (Table 2).
Compared with the FoundationOne CDx assay, both the Ion S5 XL system with the Oncomine Comprehensive assay and the Illumina NextSeq 550Dx platform with the SureSelect Custom assay successfully identified all SNVs and small indels, except for three variants (Supplemental Data Tables S1 and S2). Only one variant (ATM, c.5188C>T, VAF 2.3% in the FoundationOne CDx assay) was not called in the Oncomine Comprehensive assay. This variant was successfully detected with a 1.9% VAF in the SureSelect Custom assay (Supplemental Data Tables S1 and S2).
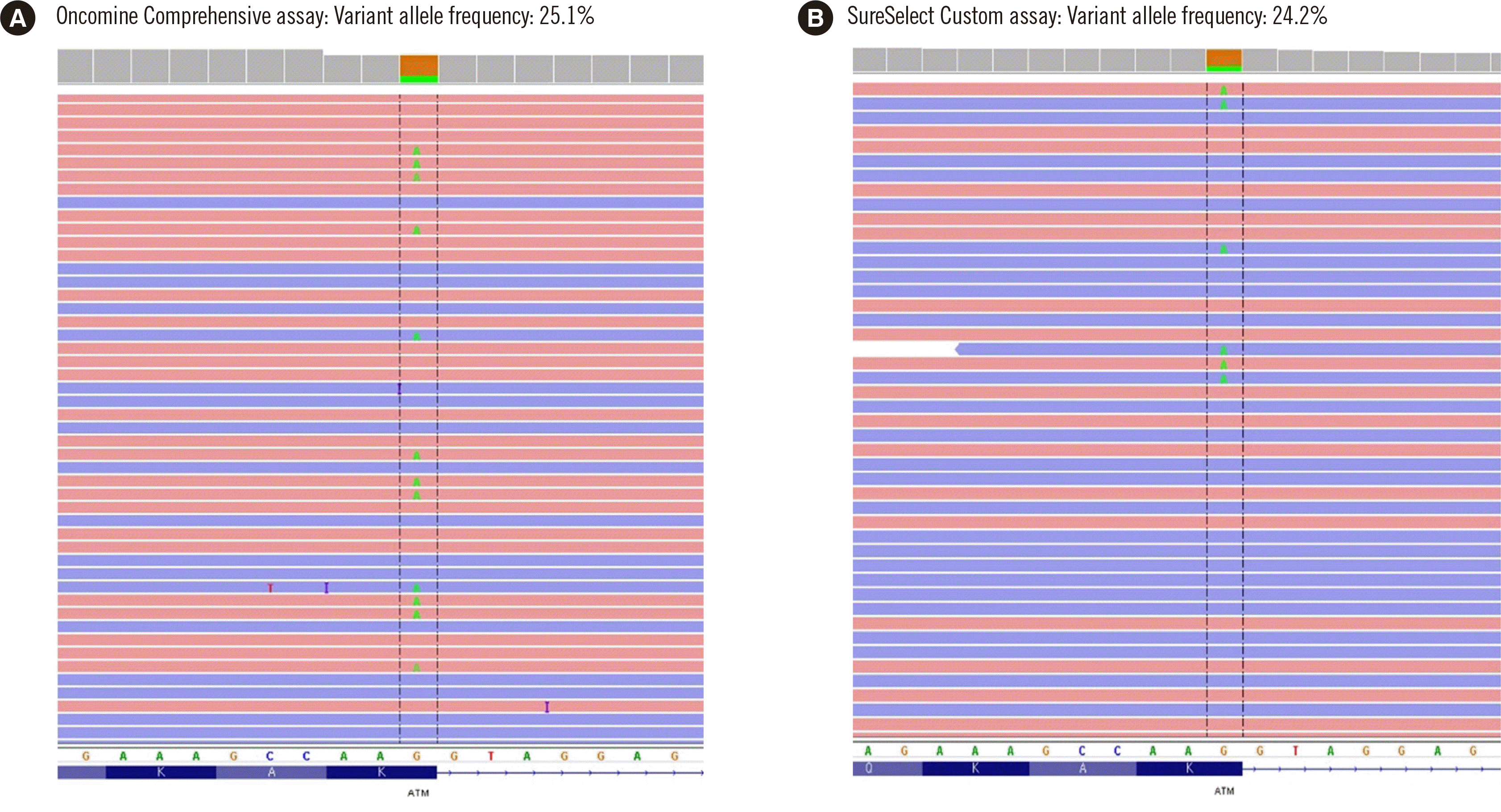
A synonymous variant with a VAF of 24.2%–25.1% [ATM, c.2250G>A, p.(Lys750=)] at the splicing junction was successfully called using both the Oncomine Comprehensive and SureSelect Custom assays. Despite being pathogenic, this synonymous variant had been rejected in the central laboratory with the FoundationOne CDx assay according to a customized analysis pipeline (Fig. 3, Table 1).
The PPA, NPA, and OPA for CNVs were 60.0%, 100%, and 99.8%, respectively, between the FoundationOne CDx and Oncomine Comprehensive assays in 84 PROfound samples (Supplemental Data Table S3). Among the five deletions (CDK12 loss, FFPE_33; ATM loss, FFPE_34; BRCA2 loss, FFPE_40; PPP2R2A loss, FFPE_73; RAD51B loss, FFPE_82), three deletions (CDK12 loss, FFPE_33; ATM loss, FFPE_34; PPP2R2A loss, FFPE_73) were also called using the Oncomine Comprehensive assay with ExomeDepth analysis.
The PPA, NPA, and OPA for CNVs were all 100% between the FoundationOne CDx and SureSelect Custom assays with ExomeDepth analysis (Supplemental Data Table S3). The SureSelect assay successfully detected three CNVs (ATM loss, FFPE_34; BRCA2 loss, FFPE_40; PPP2R2A loss, FFPE_73) that were detected in the central laboratory from 90 PROfound samples.
Comparison of HRR mutation status in mCRPC patients
We evaluated the clinical performance of NGS assays for determining mCRPC patients with deleterious variants on HRR-related genes to assess their candidature for olaparib therapy. Compared with the FoundationOne CDx assay, both the Oncomine Comprehensive and SureSelect Custom assays successfully identified mCRPC patients harboring deleterious SNVs and small indels. Two CNVs (BRCA2 loss, FFPE_40; RAD51B loss, FFPE_82) could not be detected by the Oncomine Comprehensive assay with ExomeDepth. The Oncomine Comprehensive and SureSelect Custom assays showed an OPA of 95.8% and 100%, respectively, for HRR mutation status, compared with the FoundationOne CDx assay (Table 3). Among the HRRm samples, two did not contain a qualifying HRR genomic variant other than RAD54L rearrangement or RAD51B rearrangement, which are out of the analytical range of both the Oncomine Comprehensive and SureSelect Custom assays and are not included in Table 3.
DISCUSSION
NGS is a promising tool for identifying HRR mutations in patients with mCRPC. The Illumina NGS system and Thermo Fisher Scientific’s Ion Torrent sequencing platforms, as mainstream NGS platforms, are widely used for genetic testing [12]. Illumina’s NextSeq/MiSeq platform and Thermo Fisher Scientific’s Ion S5 XL system are the main NGS equipment currently used in Korean clinical laboratories [13]. For library preparation, amplification-based and hybrid capture-based methods are primarily conducted on Illumina and Thermo Fisher Scientific sequencing platforms, respectively. Therefore, we designate the Oncomine Comprehensive assay and SureSelect Custom assay as an amplification-based library preparation NGS kit and hybridization capture-based NGS kit, respectively.
In agreement analysis, the PPA values for SNVs and indels between the central laboratory (FoundationOne CDx assay) and local laboratory (Oncomine Comprehensive assay and SureSelect Custom assay) were 98.7%–100.0%. The NPA and OPA for SNVs and indels between central and local laboratories were all 100%. Compared with that of the FoundationOne CDx assay, the OPA for CNVs of the local laboratory assays reached 99.8–100%. Both the Oncomine Comprehensive and SureSelect Custom assays successfully identified most of the mCRPC patients harboring deleterious genetic variants. The NGS approach at a local laboratory shows comparable analytical performance for defining HRR mutation status compared with the FoundationOne CDx assay approach at the central laboratory.
One variant reported by the FoundationOne CDx assay (ATM, c.5188C>T, FFPE_23, VAF 2.3%) was rejected in the Oncomine Comprehensive assay, whereas a known pathogenic variant [ATM, c.2250G>A, p.(Lys750=)] was successfully called in the Oncomine Comprehensive and SureSelect Custom assays with a VAF of 24.2%–25.1% but was not detected in the FoundationOne CDx assay. This difference is due to the bioinformatics pipeline’s variant calling strategy that filters out synonymous variants. Since pathogenic HRR mutations were not detected by all NGS assays, it is necessary to assess the possibility of false-negative or false-positive results depending on the analytical performance of the NGS platform and the final variant calling strategy.
Ion Reporter software showed that the PPA values for CNVs of the Oncomine Comprehensive assay reached 100% (95% CI: 56.6%–100%). All five deletions (CDK12 loss, FFPE_33; ATM loss, FFPE_34; BRCA2 loss, FFPE_40; PPP2R2A loss, FFPE_73; RAD51B loss, FFPE_82) detected in the central laboratory from 84 PROfound samples were also called using the Oncomine Comprehensive assay. However, false-positive CNV calls were frequently observed, and the NPA and OPA for CNVs were 92.5% (95% CI: 90.9%–93.8%) and 92.5% (95% CI: 91.0%–93.9%), respectively, between the FoundationOne CDx assay and Oncomine Comprehensive assay. The hybrid capture-based method is known to be superior to the commonly applied amplification-based methods for CNV analysis [14]. Considering the inherent limitations of the Oncomine Comprehensive assay, which is an amplification-based method, an Ion Reporter Oncomine workflow that automatically evaluates CNV calls will produce a relatively high number of false-positive results. We used ExomeDepth as an alternative method for CNV calling and comparison between platforms [9]. The PPA, NPA, and OPA for CNVs were all 100% between the FoundationOne CDx assay and SureSelect Custom assay, and were 60.0%, 100%, and 99.8%, respectively, between the FoundationOne CDx assay and Oncomine Comprehensive assay.
There are some limitations to this study. First, the proper DNA extraction of FFPE tissue samples is crucial for somatic NGS panel testing. We used extracted DNA samples provided by AstraZeneca Biobank. When evaluating the quality and concentration of DNA, some of the DNA samples did not pass the QC threshold of the input DNA sample. We considered that this was due to the lack of FFPE samples (e.g., metastatic biopsy) remaining after being subjected to NGS at Foundation Medicine, Inc. Although most pathogenic variants were successfully called and VAFs for all the pathogenic variants were highly consistent between the central and local laboratories, we could not exclude the bias of results due to the use of DNA from the same tissue blocks extracted at different times.
Second, the multigene panel NGS assay for detecting HRR mutations has not been approved as a companion diagnostic tool in Korea. We demonstrated good agreement between the FDA-approved FoundationOne CDx and locally performed NGS assays in Korea using 100 PROfound samples, but we could not experimentally define thresholds at which to call SNVs/indels and CNVs with confidence to identify patients who may benefit from treatment with the HRR-deficiency targeted therapies on each NGS platform. When a laboratory adopts multigene panel NGS assays for the HRR pathway, additional systematic validation involving wet- and dry-bench methods should be performed to determine the most appropriate cut-off values of SNVs, indels, and CNVs. Among the PROfound samples, the number of CNVs was too low to evaluate the analytical performance of detecting CNVs. Before adopting the pipeline for CNV analysis at clinical laboratories, it should be further evaluated using high-quality true-positive CNV datasets and adjusted by reviewing the false-negative and false-positive results.
There is a pressing need for comparative analytical performance data between the FDA-approved FoundationOne CDx NGS assay (central laboratory) and NGS assays of other manufacturers (local laboratory); to the best of our knowledge, studies comparing their diagnostic performances are lacking. We performed the validation of locally adopted NGS assays using samples from the PROfound trial. The HRRm cases (N=60) included the most clinically relevant HRR genes, such as BRCA1, BRCA2, ATM, PALB2, CHEK2, and other DNA repair genes that are known to be altered in mCRPC [4, 15]. The homologous recombination DDR deficiency found in these HRRm cases has been observed with a frequency >10% in several other cancers, including endometrial, biliary tract, bladder, gastroesophageal, ovarian, breast, and pancreatic cancers [16]. These data should be useful for evaluating HRR mutations in various carcinomas in the future.
 XML Download
XML Download