

 PDF
PDF Citation
Citation Print
Print



Introduction
Materials and Methods
1. Patients, clinical genetic counseling, and BRCA1/2 genotyping
2. In silico predictions and variant selection
Table 1
| Gene | HGVS name | Exon | rs ID | GnomAD frequencya) | Carrier number in our cohort | Splice predictions | |||
|---|---|---|---|---|---|---|---|---|---|
| ADA | RF | VarSeak | LaBranchoR, RNABP | ||||||
| BRCA1 | LRG_292t1:c.4484+4dupA | Ex-14 | NA | NA | 2/3,568 | NA | NA | 5b) | NA |
| BRCA1 | LRG_292t1:c.5407-10G>A | Ex-23 | rs273901767 | 4×10−6 | 2/3,568 | 0.999b) | 0.912b) | 5b) | NA |
| BRCA1 | LRG_292t1:c.4358-31A>C | Ex-14 | rs764503776 | NA | 1/3,568 | NA | NA | 1 | High, 35%b) |
| BRCA2 | LRG_293t1:c.8487G>T | Ex-19 | NA | NA | 4/3,568 | 0.999b) | 0.998b) | 5b) | NA |
| BRCA2 | LRG_293t1:c.793G>A | Ex-09 | rs1403242422 | NA | 1/3,568 | 0.067 | 0.398 | 4b) | NA |
ADA, adaptive boosting, ensemble machine learning (scoring interval 0–1); HGVS, Human Genome Variation Society; LaBranchoR, branchpoint position site predictor; RNABP, branchpoint site predictor (probability: 0–100%); NA, not applicable; RF, random Forest, ensemble machine learning (scoring interval 0–1); VarSeak, combined score for splice prediction (scoring interval 1–5).
![]()
Table 2
| Family No. | Variant (HGVS) | Age (at tumor onset) | Tumor type | Histology | No. of family membersa) with HBOC related cancer (see details on the pedigrees) |
|---|---|---|---|---|---|
| 1 | LRG_292t1:c.4484+4dupA | 29 | Breast | Invasive ductal carcinoma; ER+; PR−; HER2−; Ki67: 20% | 1 |
| 2 | LRG_292t1:c.4484+4dupA | 44 | Ovarian | Ovarian serous papillary carcinoma, high grade; multiplex metastases | 2 |
| 3 | LRG_292t1:c.4358-31A>C | 49 | Breast | Ductal carcinoma in situ; ER+; PR+; HER2+; Ki67: 10% | 5 |
| 4 | LRG_292t1:c.5407-10G>A | 48 | Breast | Invasive ductal carcinoma; ER+; PR−; HER2−; Ki67: 30% | None |
| 5 | LRG_292t1:c.5407-10G>A | 37 | Breast | Invasive ductal carcinoma; ER+; PR+; HER2−; Ki67: 80% | None |
| 6 | LRG_293t1:c.8487G>T p.(Gln2829His) | 41 | Breast | Invasive ductal carcinoma; ER+; PR−; HER2−; Ki67: 30% | 3 |
| 7 | LRG_293t1:c.8487G>T p.(Gln2829His) | 36 | Breast | Invasive ductal carcinoma; ER+; PR+; HER2−; Ki67: 15% | 3 |
| 8 | LRG_293t1:c.8487G>T p.(Gln2829His) | 45 | Breast | Invasive lobular carcinoma; ER+; PR+; HER2−; Ki67: na | 3 |
| 9 | LRG_293t1:c.8487G>T p.(Gln2829His) | 50 | Breast | Invasive ductal carcinoma; ER+; PR+; HER2−; Ki67: 15% | None |
| 10 | LRG_293t1:c.793G>A p.(Gly265Arg) | 27 | Breast | Invasive ductal carcinoma; ER−; PR−; HER2−; Ki67: 60% | 1 |
![]()
3. cDNA qualitative analyses
4. cDNA semi-quantitative measurements
5. LOH tests
6. Complex evaluation of pathogenicity
Results
1. Selection of VUS potentially affecting splicing
2. BRCA1 intronic variants causing partial exon 14 skipping
Fig. 1
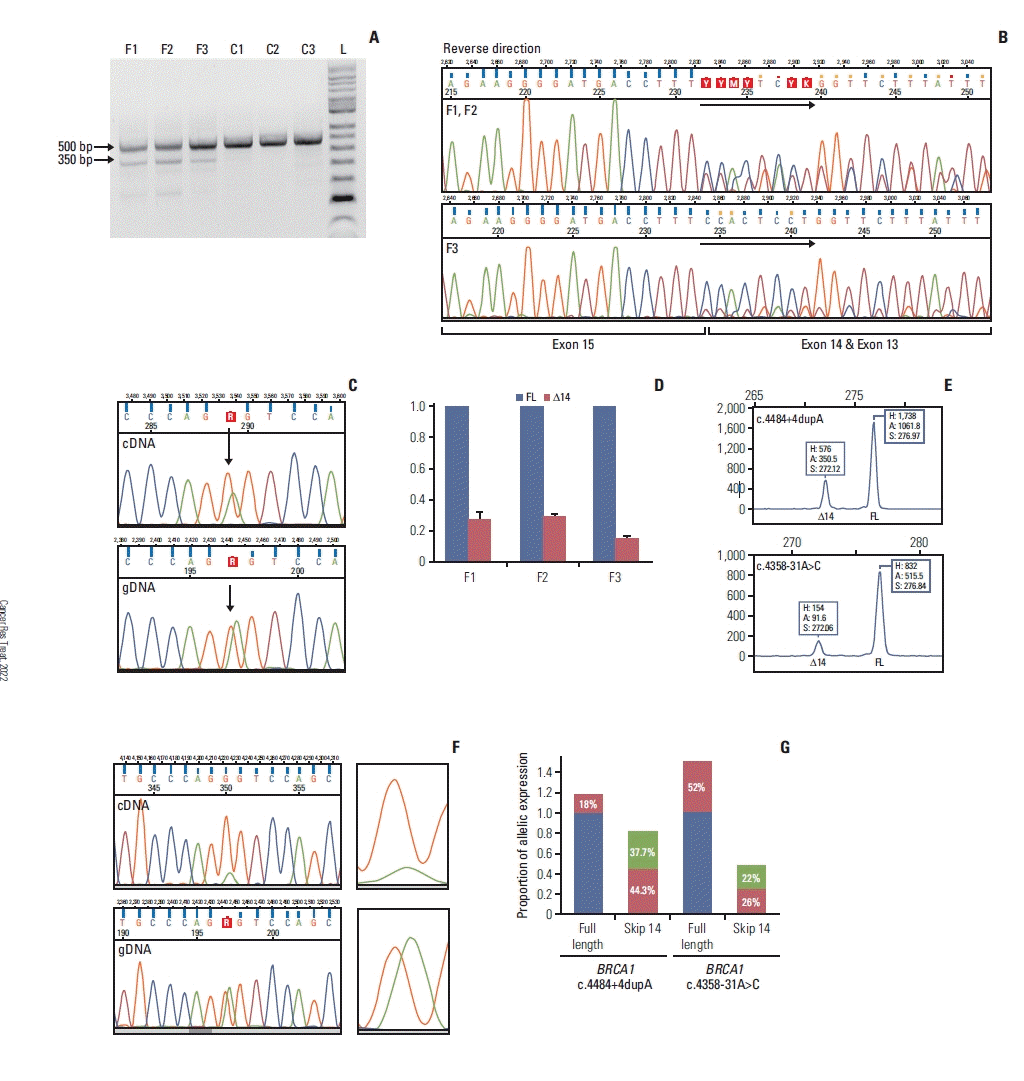
![]()
3. BRCA1 c.5407-10G>A causes partial intron inclusion
Fig. 2
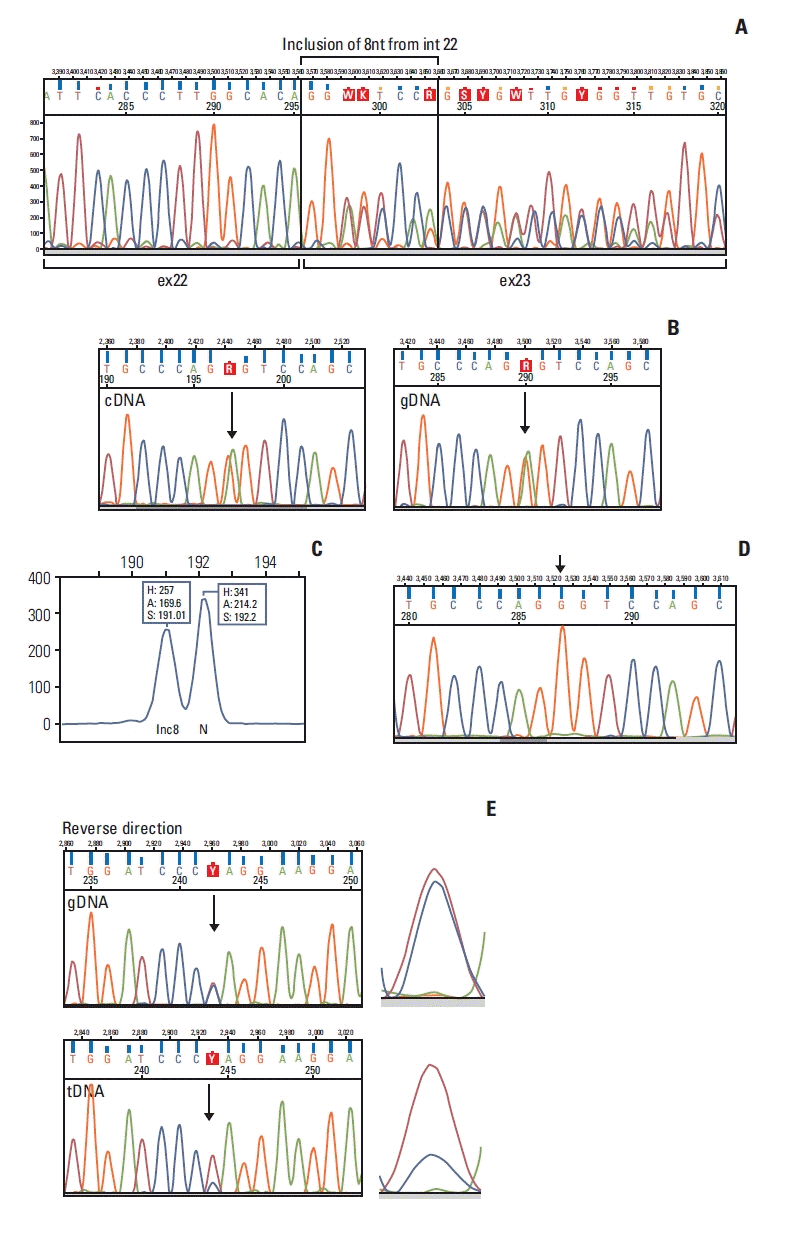
![]()
4. Transcript-level study of BRCA2 putatively spliceogenic exonic variants
Fig. 3
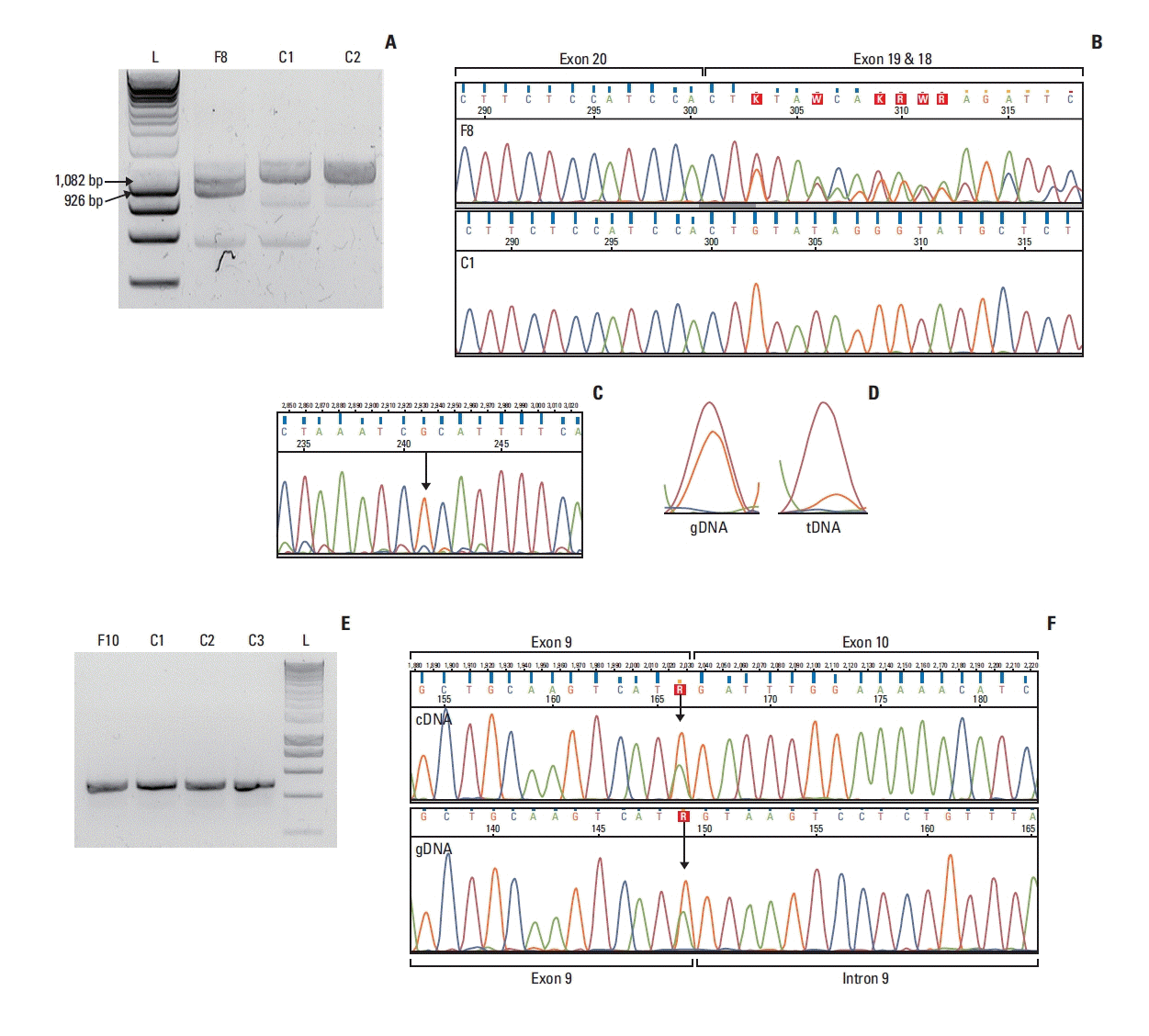
![]()
Discussion
Table 3
| Variant | ACMG class | ACMG criteria | BRCA exchange | ClinVar | ClinVar Review status | Reclassification | ACMG criteria with our evidence | Further supporting evidence |
|---|---|---|---|---|---|---|---|---|
| BRCA1: c.4484+4dupA | VUS | PM2, PP3, PP4 | No variant listed | No data | NA | LP | PM2, PP3, PP4, PS3a) | |
| BRCA1: c.5407-10G>A | VUS | PM2, PP4 | Variant not interpreted | VUS | b) | LP | PM2, PP4, PS3a) | LOH in tumor, Saturation mutagenesis Findlay et al. [23] |
| BRCA1: c.4358-31A>C | VUS | PM2, PP4, BP7 | Variant not interpreted | No data | NA | Strong VUS | PM2, PP4, BP7, PS3a) | |
| BRCA2: c.8487G>T (p.Gln2829His) | LP | PVS1, PM2, PP3, PP4, BP1 | No variant listed | NA | NA | P | PVS1, PM2, PP3, PP4, BP1, PS3a) | LOH in tumor, Aberrant splicing was formerly detected by Houdayer et al. [24]. |
| BRCA2: c.793G>A (p.Gly265Arg) | VUS | PVS1, PM2, PP4, BP1, BP4 | Not interpreted | Conflicting interpretations of pathogenicity: VUS (3) – LB (1) | b) | LB | PM2, PP4, BP1, BP4 |
PVS1: Null variant (nonsense, frameshift, canonical ±1 or 2 splice sites, initiation codon, single or multiexon deletion) in a gene where LOF is a known mechanism of disease; PM2: Variant not found in gnomAD exomes; PP3: Pathogenic computational verdict based on 1 pathogenic prediction from GERP vs. no benign predictions; PP4: Patient’s phenotype or family history is highly specific for a disease with a single genetic etiology; PS3: Well-established in vitro or in vivo functional studies supportive of a damaging effect on the gene or gene product; BP1: Missense variant in a gene for which primarily truncating variants are known to cause disease; BP4: Multiple lines of computational evidence suggest no impact on gene or gene product (conservation, evolutionary, splicing impact, etc.); BP6: Reputable source recently reports variant as benign, but the evidence is not available to the laboratory to perform an independent evaluation; BP7: A synonymous (silent) variant for which splicing prediction algorithms predict no impact on the splice consensus sequence nor the creation of a new splice site and the nucleotide is not highly conserved. ACMG, American College of Medical Genetics and Genomics; LB, likely benign; LOH, loss of heterozygosity; LP, likely pathogenic; NA, not applicable; P, pathogenic; VUS, variant of unknown significance.
![]()
 XML Download
XML Download