

 PDF
PDF Citation
Citation Print
Print



Introduction
Cancer initiation and progression involve a multistep process. Cancer initiation requires germline mutation, amplified oncogene, mutated suppressor genes, or hormone action, while cancer progression and metastasis require various growth factors, including epidermal growth factor (EGF), hepatocyte growth factor (HGF), and vascular endothelial growth factors (VEGFs), as well as proteases and adhesion molecules (Table 1) [1-6]. Cell surface receptors can bind growth factors and other ligands, which activate the receptors and transduce the signals by activating a tyrosine kinase inhibitor, thereby regulating cell functions such as cell survival, cell proliferation, protein synthesis, and angiogenesis [7].
HGF, an effector on cells expressing the N-methyl-N’-nitroso-guanidine human osteosarcoma transforming gene (c-MET) tyrosine kinase receptor, is produced by mesenchymal cells and acts on cells of epithelial origin in paracrine or autonomic fashion [8]. Studies have shown that overexpression or over-activation of HGF can lead to misplaced or inappropriately timed angiogenic and mitogenic signals. c-MET is a cell surface membrane receptor composed of a 50 kDa α-chain and a 145 kDa β-chain [9]. MET activity is observed during embryogenesis and organogenesis in normal cells and is also activated in degenerative diseases such lung and renal fibrosis and liver cirrhosis [10]. Although the HGF/c-MET axis plays a principal role in normal cell development, aberrant activation of this axis is thought to be involved in cell invasion and metastasis in most types of human cancers [11]. We have studied the HGF/c-MET pathway and the associated tumor invasion and proliferation in gastric cancer for several years and here, we review our experiment results.
Interaction of HGF and other molecular proteins in gastric cancer
1. E-cadherin and β-catenin
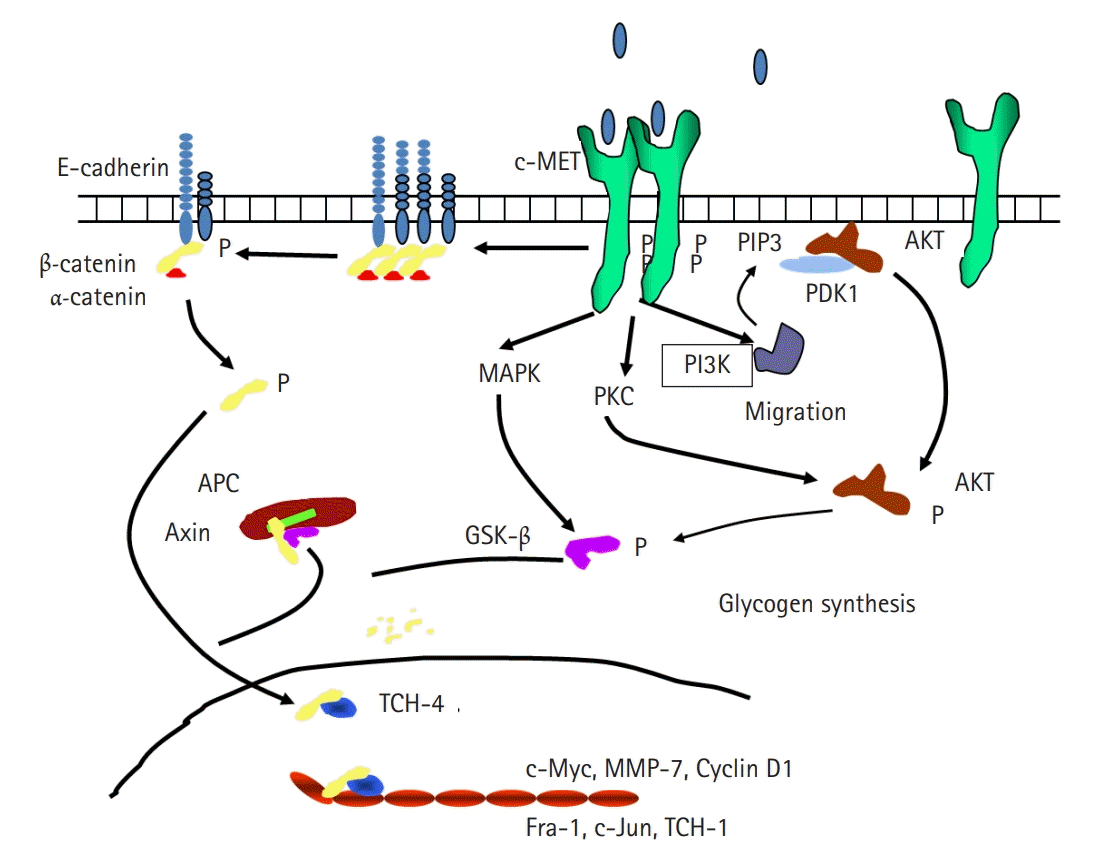
MET has been demonstrated to interact with other cell surface receptors, including integrins, human epidermal growth factor receptor, and FAS receptor, to enhance downstream signaling and tumorigenesis. We previously reported that the expression of E-cadherin (ECD) and urokinase plasminogen activator (uPA) is associated with the development of pancreatic cancer [12]. ECD is a transmembrane glycoprotein that is responsible for calcium-dependent intercellular adhesion by homotypic interaction and is one of the principal elements of the cytoskeleton. Decrease or loss of ECD is frequently associated with cell-to-cell disengagement, tumor invasion, and metastasis [13,14]. ECD functions to dephosphorylate β-catenin, thus inhibiting the binding of intracellular ECD to catenin proteins. It has been suggested that HGF reduces cell-to-cell adhesion by dephosphorylation of the ECD/catenin complex and ECD shedding [15]. We hypothesized that HGF/c-MET may interact with ECD to promote tumorigenesis through the activation of matrix metalloproteinase-7 (MMP-7), which degrades many cellular matrix proteins and adhesion molecules (Fig. 1). To confirm this hypothesis, we investigated the association between HGF/c-MET, ECD, and MMP-7 in two stomach cancer cell lines, NUGC-3 and MKN-28. Western blot and reverse transcription PCR analyses showed that treatment of these cells with HGF reduced the expression of ECD. These results suggested that HGF may stimulate the extracellular cleavage of ECD, thereby increasing the shedding of the soluble fragment and decreasing the 120-kDa full-length ECD in the total cell lysates [16]. MMP-7, which is known to be expressed predominantly by tumor cells in various cancers, was increased by HGF treatment and knockdown of MMP-7 expression in the stomach cancer cell lines resulted in no extracellular cleavage of ECD as well as decreased in vitro cell invasion. These results suggest that HGF may interact with ECD, leading to the activation of the MMP-7 pathway and increased cell invasion.
2. Urokinase plasminogen activator
uPA is a member of the family of serine proteases and is known to participate in cell migration and tissue remodeling. uPA overexpression has been reported in lung, colon, and breast cancers [17-19]. Many studies have shown that blocking the expression of uPA or inhibiting its binding to the uPA receptor (uPAR) suppresses tumor cell invasion and metastasis in various cancer cell lines [20,21]. We measured uPAR expression in 26 patients with stomach cancer before and after surgery and found that uPAR expression was significantly decreased after surgery (p<0.05). We also found that the survival rate of patients with gastric tumors expressing uPAR was significantly lower than that of patients with tumors not expressing uPAR (p=0.035) [22]. We hypothesized that uPA is also associated with tumor progression in gastric cancer and we explored the relationship between HGF and uPA in gastric cancer tumorigenesis. We found that HGF induced reactive oxygen species generation, which regulates uPA production and tumor invasion via mitogen-activated protein (MAP) kinase [23]. Previous studies have also examined the connection between HGF and uPA. One study showed that histone deacetylase (HDAC) regulates HGF-induced expression of both uPA and MMP-9 through a protein kinase C (PKC) dependent pathway in gastric cancer [24]. Another study showed that survivin, a member of the inhibitor of apoptosis family, increases HGF-induced uPA expression and seems to play a role in gastric cancer tumorigenesis [25].
3. New HGF regulatory genes
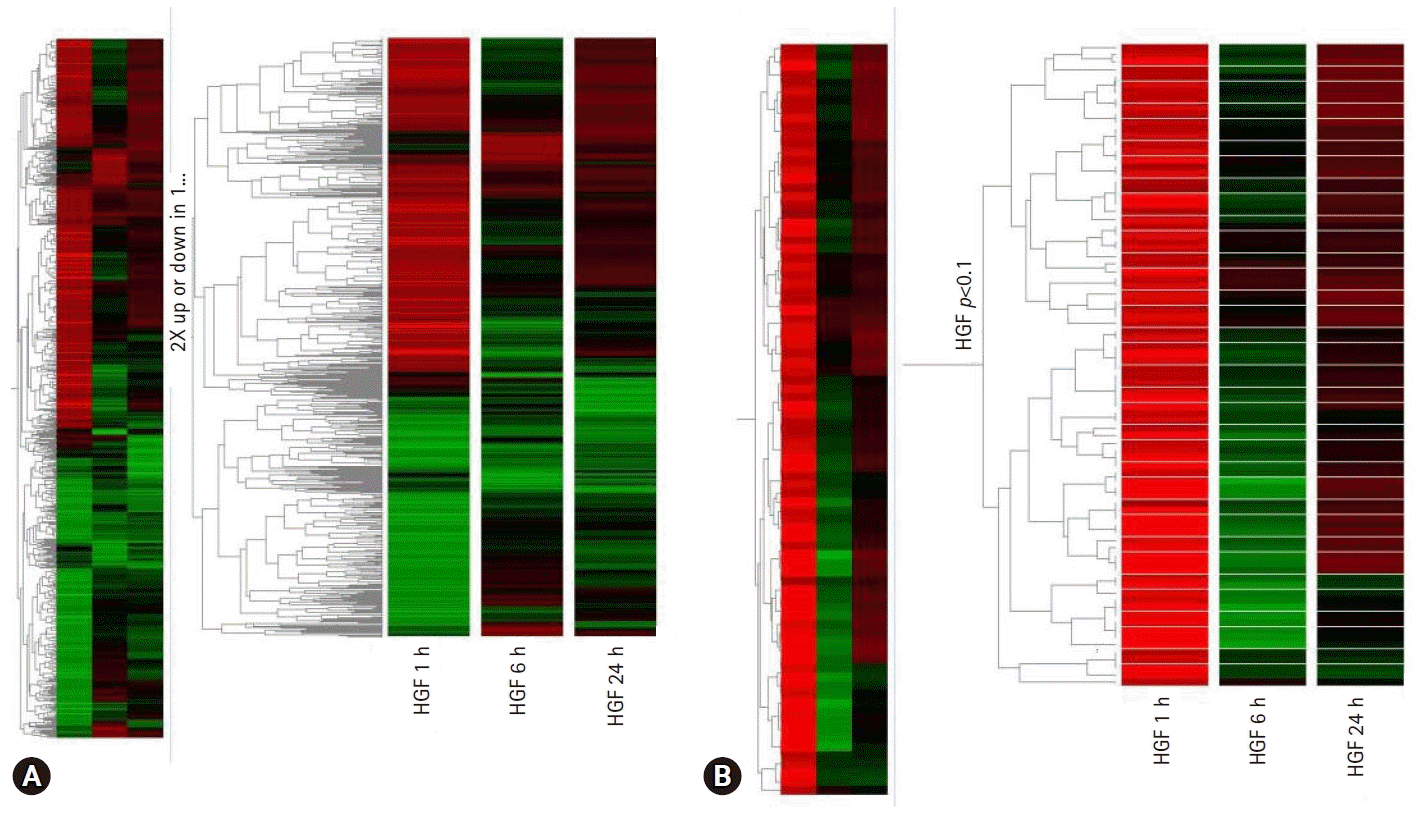
To find new HGF regulatory genes and identify their role in HGF-induced stomach cancer cell survival, we screened for genes induced by HGF using complimentary deoxyribonucleic acid (cDNA) microarray technology in the stomach cancer cell lines, NUGC-3 and MKN-28 (Fig. 2). We selected the genes that were up or downregulated by more than three-fold in NUGC-3 and MKN-28 cells during HGF treatment (Table 2) and determined their function in conjunction with HGF.
Bcl-2 associated agonist of cell death (BAD), a BH3-only proapoptotic Bcl-2 family protein, has been found to be upregulated in response to HGF treatment. BAD functions by inactivating anti-apoptotic Bcl-2 proteins [26]. cDNA microarray analysis results have confirmed that BAD is upregulated at the RNA and protein levels following HGF treatment. Our data showed that HGF induced BAD overexpression and enhanced BAD phosphorylation, thereby inhibiting apoptosis and promoting cancer cell survival [27].
KiSS-1 was also upregulated in response to HGF treatment. KiSS-1 is a putative metastasis suppressor gene and its expression is increased in several human malignancies including melanoma [28] and breast cancer [29]. One study also reported that overexpression of KiSS-1 in breast cancer cells results in a more aggressive phenotype [30]. Consistent with these results, we found that HGF induced the overexpression of KiSS-1 in a p38-dependent manner. In addition, KiSS-1 suppressed MMP-9 expression and decreased cell invasion in vitro, suggesting it may act as a metastasis suppressor gene in gastric cancer [31].
Jun B was also upregulated in response to HGF treatment. Jun B belongs to the June gene family (c-Jun, JunB, and JunD), whose members encode the activator protein-1 (AP-1) family of transcription factors. AP-1 is a dimeric transcription factor that is enhanced by the MAP kinase pathway in the presence of growth factors, hormones, or other environmental stresses [32,33]. Of the AP-1 components, c-Jun and c-Fos were first identified as viral oncoproteins; thus, their function in tumorigenesis has been established. However, it is also known that some Jun and Fos proteins can suppress tumor formation [34]. Accordingly, we examined the role of Jun B in gastric cancer. In our study, Jun B levels were decreased by inhibition of nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB), and cell proliferation and invasion were decreased in Jun B knockdown stomach cancer cell lines. Further, Jun B knockdown cells blocked the MMP-9 upregulation induced by HGF. MMP-9 is a matrix metalloproteinase protein that degrades the basement membrane, exposing cryptic sites within the matrix and resulting in cancer cell invasion [35,36]. These data suggest that Jun B expression induced by HGF can activate MMP-9 by the NF-κB pathway and thereby contribute to invasion and cell proliferation in gastric cancer [37].
We recently studied that lipocalin-2 (LCN2) is upregulated by HGF treatment. LCN2 is a member of the lipocalin family, which binds and transports small lipophilic molecules including leukotrienes, retinoic acids, and prostaglandins, and it was first identified as a modulator of the immune system [38]. In addition, LCN2 binds MMP-9, forming a complex comprising LCN2 and MMP-9, promoting MMP-9 activation, and preventing its degradation [35,36]. HGF treatment upregulated the expression of LCN2 in gastric cancer cells, leading to increased activation of MMP-9. Knockdown of LCN2 in these cells decreased MMP-9 activation in response to HGF treatment and treatment of the cells with an NF-κB inhibitor prevented the HGF-mediated upregulation in LCN2 expression. Further, HGF-mediated cell proliferation and invasion was decreased in LCN2 knockdown cells compared to control cells [39]. These data suggest that HGF induces the upregulation of LCN2 expression, which activates MMP-9, and HGF may play a role in proliferation and invasion of gastric cancer.
Conclusion
Abberant activation of MET signaling occurs in a subset of advanced cancers, including gastric cancer. The HGF/c-MET axis interacts with several molecules including ECD, uPA, KiSS-9, Jun B, and LCN2. This pathway may affect cell invasion and metastasis or cell apoptosis and is therefore associated with tumorigenesis and metastasis in gastric cancer, which maybe one of the important therapeutic targets. To validate our findings, further experiments are warranted using in vivo knockout mouse models.
 XML Download
XML Download