

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Chronic kidney disease (CKD) is the gradual loss of kidney function accompanied by kidney damage or a decreased glomerular filtration rate over a period of months to years. Patients with CKD, particularly those undergoing dialysis, have various complications, such as cardiovascular and mineral bone disorders. Patients with CKD also show reduced muscle mass and exercise capacity [1]. Therefore, muscle mass is considered an indicator of the clinical condition of patients with CKD [2]. Sarcopenia, which is defined as the loss of skeletal muscle function, because of decreased muscle mass and strength [3], is highly prevalent in patients with CKD [4] and is a serious problem, as it increases mortality and morbidity [5]. Therefore, maintaining muscle mass and strength is important for improving the prognosis of patients with CKD. Restriction of salts and proteins is a fundamental dietary therapy for CKD patients; in contrast, protein supplementation is a fundamental dietary therapy for patients with sarcopenia [67]. Because conflicting dietary therapies are recommended for CKD and sarcopenia, nutritional management has not been well established for individuals with both these complications.
Exercise capacity depends on many factors such as cardiopulmonary function, muscle mass, and mitochondrial function [8910]. Muscle atrophy in patients with CKD associated with mitochondrial dysfunction [1112]. Mitochondrial DNA (mtDNA) copy number, an indicator of mitochondrial content, is lower in patients with CKD than in healthy subjects. Furthermore, a decreased mtDNA copy number is a potential risk factor for mortality in patients with CKD [12]. Therefore, prevention and amelioration of mitochondrial dysfunction is important for improving muscle atrophy and decreasing the risk of mortality in patients with CKD.
Patients with CKD have a high concentration of uremic toxins in their blood. Indoxyl sulfate (IS) is a uremic toxin produced by metabolism of tryptophan in a diet. IS accumulates in the blood of patients with CKD and impairs the functioning of the liver, thyroid gland, and kidneys [1314]. IS can cause muscle atrophy in CKD mice through mitochondrial dysfunction [15]. However, the mechanisms underlying IS-induced mitochondrial dysfunction in muscles have not been well elucidated.
Ursolic acid (UA) is a natural triterpene compound found in various fruits and vegetables, such as apples and olives, respectively [16]. UA has a variety of physiological activities, including anti-hyperglycemic [17], anti-inflammation [18], and anti-cancer [19] activities. In addition, UA has shown exercise-mimicking effects, such as promoting muscle hypertrophy and improving insulin sensitivity, in mice [20]. In humans, oral administration of UA remarkably promotes an increase in muscle strength by resistance training [21]. Although the physiological effects of UA on muscles have been reported in healthy humans and rodents, the effects of UA on muscular atrophy in chronic diseases, such as CKD, have not been well reported. Therefore, the aim of this study was to evaluate the physiological effects of UA on mitochondrial biogenesis in CKD.
Go to : 
MATERIALS AND METHODS
Cell culture
C2C12 cells, derived from an immortalized mouse myoblast cell line, were obtained from the Riken Cell Bank (Tsukuba, Japan). The cells were cultured in Dulbecco's modified Eagle's medium (DMEM; FUJIFILM Wako Pure Chemical, Osaka, Japan) containing 10% fetal bovine serum (Biowest, Nuaille, France) and 1% penicillin-streptomycin (FUJIFILM Wako Pure Chemical) in a humid environment containing 5% CO2 at 37°C. The cells were seeded at a density of 7.5 × 105/φ35 mm dish. To promote differentiation, the medium was replaced with DMEM containing 2% horse serum (Sigma-Aldrich, St. Louis, MO, USA) after the cells reached 90–100% confluence. Then, the cells were treated with or without IS (IS potassium salt; Nacalai Tesque, Kyoto, Japan; 0.1 mM) and UA (Tokyo Kasei Kogyo Co., Tokyo, Japan; 1 or 2 μM) for 6 days. The differentiation medium was replaced every second days.
Cell viability
The effects of IS and UA on cell viability were measured using a neutral red assay [22]. A neutral red stock solution (0.4% neutral red in water) was diluted to 1:80 (v/v) with phosphate buffered saline (PBS; Nissui Pharmaceutical, Tokyo, Japan). C2C12 cells were treated with or without IS and/or UA for 6 days. At the end of the incubation period, the cells were incubated with the neutral red solution at 37°C for 30 min. Neutral red was extracted from the cells by adding a mixture of 1% (w/v) acetic acid and 50% (v/v) ethanol, followed by culturing at 20–25°C for 30 min. Finally, absorbance was measured in a microplate reader at 540 nm.
Reverse transcription and real-time quantitative polymerase chain reaction (PCR)
Total RNA was extracted from C2C12 cells using ISOSPIN Cell & Tissue RNA (Nippon Gene, Tokyo, Japan) according to the manufacturer's instructions. After the RNA concentration was measured, equal amounts of total RNA were reverse-transcribed into cDNA using a PrimeScript™ RT reagent kit (TaKaRa Bio, Shiga, Japan) as per the manufacturer's instructions. SYBR green-based real-time PCR was performed in 96-well plates using TB Green™ Premix Ex Taq II reagents (TaKaRa Bio) and StepOnePlus (Thermo Fisher Scientific, Waltham, MA, USA). The reaction procedure was as follows: 95°C for 30 s, followed by 35 cycles at 95°C for 5 s and 60°C for 30 s. The specificity of the real-time PCR product was demonstrated by melting curve analysis and agarose gel electrophoresis. Relative mRNA expression levels were normalized to that of β-actin. The data were obtained using the ΔΔCt method. The primer sequences are listed in Table 1.

Table 1
Primer sequences used in real-time PCR analysis
PCR, polymerase chain reaction; Myod1, myogenic differentiation 1; MyoG, myogenin; Mfn1, mitofusin 1; Mfn2, mitofusin 2; Drp1, dynamin 1-like; Fis1, fission, mitochondrial 1; Ppargc1a, peroxisome proliferative activated receptor, gamma, coactivator 1 alpha; Nrf1, nuclear respiratory factor 1; Tfam, transcription factor A, mitochondrial; Sirt1, sirtuin 1; Mef2c, myocyte enhancer factor 2C; IL-6, interleukin 6; Pdk4, pyruvate dehydrogenase kinase, isoenzyme 4; Acadm, acyl-Coenzyme A dehydrogenase, medium chain; Cpt1b, carnitine palmitoyltransferase 1b; Cd36, CD36 molecule; Cycs, cytochrome c, somatic; Atp5b, ATP synthase, H+ transporting mitochondrial F1 complex, beta subunit; ND1, NAD(P)H dehydrogenase 1; b-actin, beta-actin.
![]()
Western blotting
Cells were rinsed with cold PBS and lysed with radioimmunoprecipitation assay buffer containing protease and phosphatase inhibitor cocktails (Nacalai Tesque). Equal amounts of proteins were electrophoresed in 10% sodium dodecyl sulphate-polyacrylamide gels and transferred to a polyvinylidene difluoride membrane using a semi-dry blotting apparatus. Membranes were blocked with 1% skimmed milk in Tris-buffered saline-Tween overnight and incubated with primary antibodies (Cell Signaling Technology, Danvers, MA, USA; anti-phospho-extracellular signal-regulated kinase (ERK) 1/2, anti-ERK1/2, anti-phospho-AKT, anti-AKT, anti-phospho-STAT3, anti-STAT3, and β-actin) for 2 h, followed by incubation with an HRP peroxidase-conjugated secondary antibody (Cell Signaling Technology) for 1 h. After several washing steps, immunodetection was performed using EzWestLumi plus (ATTO Co., Tokyo, Japan).
mtDNA copy number
Total DNA was extracted from C2C12 cells using a NucleoSpin Tissue Kit (Macherey-Nagel, Düren, Germany) according to the manufacturer's instructions. The mtDNA copy number was defined as mtDNA (ND1): nuclear DNA (b-actin), and was determined by real-time PCR, as described in the section “Reverse transcription and real-time quantitative polymerase chain reaction (PCR).”
ATP levels
C2C12 cells were seeded at a density of 7.5 ×105 cells/well in a 96-well plate. After the cells grew to 90%~100% confluence, they were incubated with or without IS (0.1 mM) and UA (1 or 2 μM) for 6 days. Following incubation, ATP levels were measured using the CellTiter-Glo® 2.0 assay (Promega, Madison, WI, USA) according to the manufacturer's instructions.
Interleukin (IL)-6 secretion measurements
After C2C12 cells were incubated with or without IS (0.1 mM) and UA (1 or 2 μM) for 4 days, the culture medium was collected. The concentration of IL-6 was measured by enzyme-linked immunosorbent assay (ELISA) using a mouse IL-6 uncoated ELISA kit (Thermo Fisher Scientific) according to the manufacturer's instructions.
Statistical analysis
Data are presented as the means ± SD of 3 or 6 independent experiments. Comparisons between multiple groups were performed using one-way analysis of variance followed by Dunnett's multiple comparison post hoc test. P < 0.05 was considered significant. Statistical analysis was performed using Statcel-4 (OMS Inc., Tokorozawa, Japan).
Go to :
RESULTS
Effects of IS and UA on the cell viability and differentiation of C2C12 cells
The IS concentration increases up to 0.2 mM in the blood of patients with CKD [23]. A recent study reported that IS decreased the viability of C2C12 cells at pharmacological concentrations (> 1 mM) [24]. However, it is unclear whether IS decreases cell viability at physiological concentrations in patients with CKD. As shown in Fig. 1A, there was no significant difference in cell viability between the groups. To examine the differentiation state of the C2C12 cells, we measured the mRNA levels of Myod1 and Myog, which are well-known differentiation markers [25]. As shown in Fig. 1B, the mRNA expression levels of Myod1 and Myog were significantly decreased after treatment with IS; however, this decrease was significantly improved after treatment with UA. Thus, IS suppressed the differentiation of C2C12 cells without decreasing cell viability, and this suppression was ameliorated by treatment with UA.
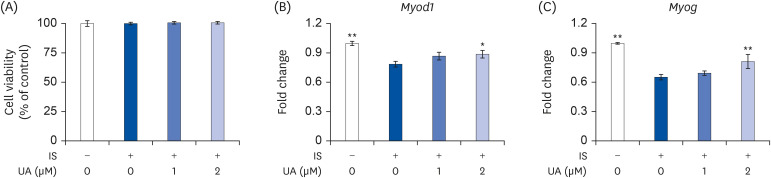 | Fig. 1Effects of IS and UA on the cell viability and differentiation of C2C12 cells. C2C12 cells were treated with or without IS (0.1 mM) and UA (1 or 2 µM) for 6 days. (A) Cell viability was assessed using a neutral red assay. (B) The mRNA expression of differentiation marker genes (Myod1 and Myog) was measured by real-time RT-PCR. Data are expressed as mean ± SD (n = 6).IS, indoxyl sulfate; UA, ursolic acid; RT-PCR, reverse transcription-polymerase chain reaction.
Each group was compared with the IS-treated group by one-way analysis of variance followed by Dunnett's test, *P < 0.05 and **P < 0.01.
|
UA improved the IS-induced reduction of ERK1/2 and AKT activities in C2C12 cells
To elucidate the mechanisms of IS and UA in the differentiation of C2C12 cells, we assessed the activities of ERK1/2 and AKT. ERK1/2 and AKT activities are stimulated by more than 100 folds upon phosphorylation of the activation loop residues Thr202/Tyr204 and Ser473, respectively [2627]. The extent of Thr202/Tyr204 and Ser473 phosphorylation indicates the ERK1/2 and AKT activities. As shown in Fig. 2, ERK1/2 and AKT activities were significantly decreased by treatment with IS; however, this decrease was improved after treatment with UA in a dose-dependent manner. Perhaps, IS and UA regulate differentiation through ERK1/2 and AKT activities in C2C12 cells.
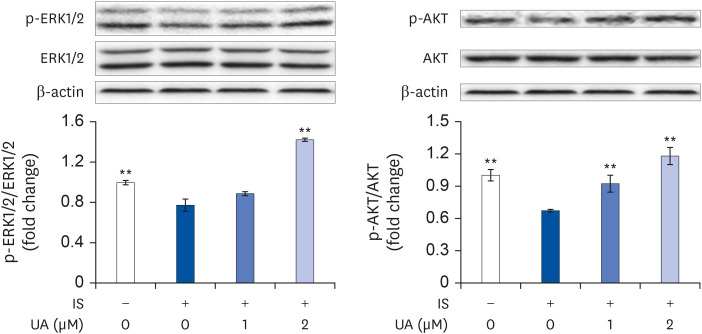 | Fig. 2Effects of IS and UA on ERK1/2 and AKT phosphorylation.Cells were treated with or without IS (0.1 mM) and UA (1 or 2 µM) for 1 h. The levels of ERK1/2, AKT, and their phosphorylated states were assessed by western blotting. b-actin was used as the internal control. Results are expressed as mean ± SD (n = 3).
IS, indoxyl sulfate; UA, ursolic acid; ERK, extracellular signal-regulated kinase; p-, phospho-.
Each group was compared with the IS-treated group by one-way analysis of variance followed by Dunnett's test, **P < 0.01.
|
UA improved the IS-induced reduction in mitochondrial contents in C2C12 cells
To assess the effects of IS and UA on the mitochondrial content in C2C12 cells, we measured the mtDNA copy number using real-time PCR. As shown in Fig. 3A, the mtDNA copy number was significantly decreased after the treatment with IS. However, UA significantly improved the IS-induced mtDNA copy number decrease in a dose-dependent manner. Mitochondria undergo fusion and fission, which comprise mitochondrial dynamics, to maintain mitochondrial quality and function after biogenesis. The mtDNA copy number is regulated by mitochondrial dynamics and biogenesis [28]. To assess the effects of IS and UA on mitochondrial dynamics, we investigated the mRNA expression levels of mitochondrial fusion (Mfn1 and Mfn2) and fission (Drp1 and Fis1) marker genes. As shown in Fig. 3B, the mRNA expression of Mfn1 and Mfn2 was significantly decreased upon the treatment with IS; however, this decrease was improved after treatment with UA. In contrast, there was no significant difference in Drp1 and Fis1 expression levels upon treatment with IS (Fig. 3C). The mRNA expression levels of mitochondrial biogenesis biomarkers (Ppargc1a, Nrf1, Tfam, Sirt1, and Mef2c) were decreased by treatment with IS; however, the decrease was improved upon treatment with UA (Fig. 3D).
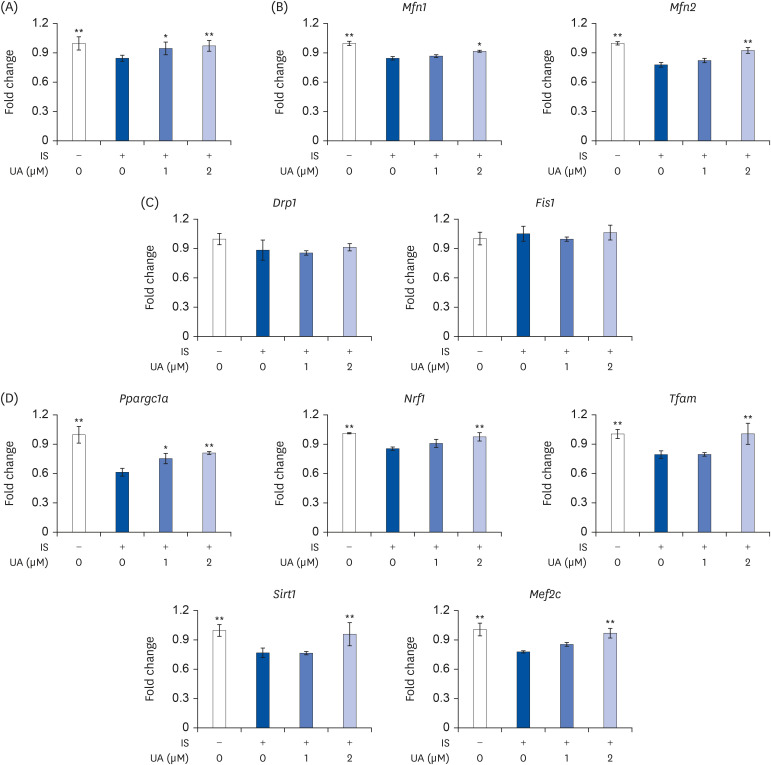 | Fig. 3Effects of IS and UA on mtDNA copy number and mitochondrial biogenesis and dynamics in C2C12 cells. C2C12 cells were treated with or without IS (0.1 mM) and UA (1 or 2 µM) for 6 days. (A) Using real-time PCR, the mtDNA copy number, defined as the ratio of mitochondrial DNA-encoded ND1 to nDNA-encoded b-actin, was determined. (B, C) The mRNA expression levels of mitochondrial fusion (Mfn1 and Mfn2) and fission (Drp1 and Fis1) marker genes were measured by real-time RT-PCR. (D) The mRNA expression levels of Ppargc1a, Nrf1, Tfam, Sirt1, and Mef2c were measured by real-time RT-PCR. Results are expressed as mean ± SD (n = 6).IS, indoxyl sulfate; UA, ursolic acid; mtDNA, mitochondrial DNA; PCR, polymerase chain reaction; nDNA, nuclear DNA; RT-PCR, reverse transcription-polymerase chain reaction.
Each group was compared with the IS-treated group by one-way analysis of variance followed by Dunnett's test, *P < 0.05 and **P < 0.01.
|
UA improved the IS-induced increase in the mRNA expression and secretion levels of IL-6
To further investigate the pathways involved in mitochondrial biogenesis, we examined IL-6 expression and secretion levels. As shown in Fig. 4, IS increased the intracellular mRNA (Fig. 4A) and secretory protein (Fig. 4B) levels of IL-6; however, this increase was significantly suppressed by treatment with UA. STAT3 activity is stimulated by the phosphorylation of its activation loop residue of Tyr705 [29]. IL-6 increases STAT3 activity to regulate mitochondrial homeostasis in C2C12 cells. Therefore, we examined the effects of IS and UA on intracellular STAT3 activity. As shown in Fig. 4C, STAT3 activity was significantly increased after treatment with IS; however, the increase improved upon treatment with UA in a dose-dependent manner. Perhaps, UA improved mitochondrial biogenesis impairment by suppressing the IS-induced increase in IL-6 levels.
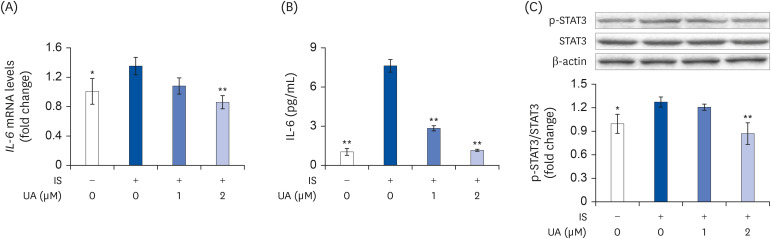 | Fig. 4Effects of IS and UA on IL-6 expression in C2C12 cells. Cells were treated with or without IS (0.1 mM) and UA (1 or 2 µM) for 4 days. (A) IL-6 mRNA expression was measured using real-time RT-PCR. (B) Secretion levels of the IL-6 protein in the culture media were measured by enzyme-linked immunosorbent assay. (C) The quantified levels of p-STAT3 were normalized to that of STAT3. b-actin was used as the internal control. Data are expressed as mean ± SD (n = 3).IS, indoxyl sulfate; UA, ursolic acid; RT-PCR, reverse transcription-polymerase chain reaction; p-, phospho-.
Each group was compared with the IS-treated group by one-way analysis of variance followed by Dunnett's test, *P < 0.05 and **P < 0.01.
|
UA improved the IS-induced reduction in ATP levels in C2C12 cells
The mRNA expression of representative fatty acid oxidation (FAO: Pdk4, Acadm, Cpt1b, and Cd36) and oxidative phosphorylation (OXPHOS: Cycs and Atp5b) genes was downregulated following treatment with IS; however, this was completely reversed with the UA treatment (Fig. 5A and B). The ATP levels decreased slightly but significantly with IS treatment; however, this was reversed with the UA treatment (Fig. 5C).
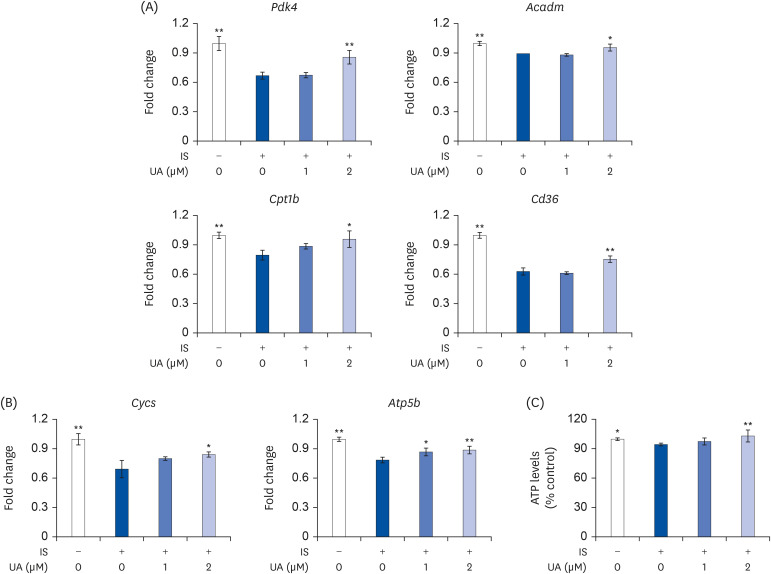 | Fig. 5Effects of IS and UA on ATP levels in C2C12 cells. Cells were treated with or without IS (0.1 mM) and UA (1 or 2 µM) for 6 days. (A, B) The mRNA expression levels of Pdk4, Acadm, Cpt1b, Cd36, Cycs, and Atp5b were measured by real-time RT-PCR. (C) The ATP levels in C2C12 cells were assessed using the CellTiter-Glo® 2.0 assay. Data are expressed as mean ± SD (n = 6).IS, indoxyl sulfate; UA, ursolic acid; RT-PCR, reverse transcription-polymerase chain reaction.
Each group was compared with the IS-treated group by one-way analysis of variance followed by Dunnett's test *P < 0.05 and **P < 0.01.
|
Go to :
DISCUSSION
In this study, we investigated the effects of IS on C2C12 cells to elucidate the mechanisms underlying the muscle dysfunction associated with CKD. This study was conducted with 0.1 mM IS, which is the physiological blood concentration in patients with CKD. Our major findings were as follows: i) IS induced impairment in myoblast differentiation, ii) IS decreased mitochondrial content and ATP levels in C2C12 cells, and iii) IS increased IL-6 secretion from C2C12 cells. In addition, we revealed that UA improved the IS-induced impairments in C2C12 cells.
Nitrogen in dietary protein is mainly metabolized to urea and then excreted in the urine. However, as the excretory function is impaired in patients with CKD, their blood contains higher concentrations of nitrogen-containing urea toxins, such as IS, urea, and creatinine; in fact, the IS concentration can increase up to 0.2 mM in the blood of patients with CKD [23]. A recent study reported that IS significantly decreased the viability of C2C12 cells a concentration of 1 mM [24]. However, we found that IS (0.1 mM) did not significantly affect the cell viability (Fig. 1A). This result suggests that cell death in muscles is not caused by the physiological concentrations of IS in patients with CKD.
Further, muscle strength and exercise capacity decrease in the early stages of CKD [30]. Muscle atrophy is a major complication of CKD and is associated with increased morbidity and mortality. Muscle atrophy is caused by the inactivation of myoblasts and inhibition of differentiation [31]. In this study, IS reduced the mRNA expression of Myod1 and Myog (Fig. 1B), which are differentiation markers, in C2C12 cells without decreasing cell viability. These results suggest that IS induced muscular atrophy by suppressing differentiation rather than by causing cell death at physiological concentrations in patients with CKD. Muscle hypertrophy is induced by oral administration of UA [20]. Intraperitoneal administration of UA has been known to promote muscle hypertrophy in combination with low-intensity exercise in rats [32]. However, to our knowledge, the effect of UA on chronic disease-related muscular atrophy has not yet been reported. In this study, UA ameliorated the IS-induced decrease in the expression of differentiation markers (Fig. 1B). These results suggest that UA may improve IS-induced muscular atrophy and ameliorate the poor prognosis of patients with CKD.
The activities of ERK1/2 and AKT play pivotal roles in the differentiation of C2C12 cells. 4-hydroxy-3-methoxy cinnamic acid promotes the differentiation of C2C12 cells through an increased expression of Myod1 and Myog. These effects are suppressed upon treatment with ERK1/2 and AKT inhibitors [30]. In this study, the activities of ERK1/2 and AKT were significantly decreased by treatment with IS; however, this decrease was improved upon treatment with UA (Fig. 2). These results suggest that ERK1/2 and AKT activities are involved in the regulation of IS and UA in the differentiation of C2C12 cells.
Mitochondria play an important role in myogenesis. As myoblasts differentiate into myotubes, the content and morphology of the mitochondria are altered. For example, mitochondrial content, mass, and volume are remarkably increased after the onset of myoblast differentiation [33]. However, in a CKD mouse model induced by mutations in the Anks6 gene, the expression levels of Myod and Myog increased despite muscle atrophy and downregulation of mitochondrial biogenesis [34]. Therefore, to evaluate CKD-related muscle atrophy, it is important to evaluate, not only myoblast differentiation, but also mitochondrial biogenesis. In this study, mtDNA copy number, which is an indicator of mitochondrial content, was decreased on treatment with IS; however, this decrease was improved after treatment with UA (Fig. 3A). Previous studies have reported that the mtDNA copy number in the muscles is reduced in patients with CKD stage 5 undergoing hemodialysis [11]. These results suggest that UA possibly suppresses the decrease in the mitochondrial content and improves muscle function in CKD patients.
Mitochondrial dynamics are important for maintaining the overall morphology of mitochondria. Disorders of mitochondrial dynamics are associated with various diseases, including cancer, cardiovascular diseases, and diabetes [3536]. However, the involvement of mitochondrial dynamics in CKD progression has not been well elucidated. As shown in Fig. 3B, we observed that the expression of mitochondrial fusion markers (Mfn1 and Mfn2) decreased upon treatment with IS; however, this decrease was improved after treatment with UA. In contrast, the expression of the fission markers (Drp1 and Fis1) was not significantly altered after treatment with IS and UA (Fig. 3C). Mitochondrial fusion is essential for maintaining the homeostasis of mtDNA, because the deficiency of mitochondrial fusion factors induces accumulation of mtDNA mutations and a reduction in the mtDNA copy number [37]. Although mitochondrial fission is not important for maintaining mtDNA levels, it is involved in the clearance of dysfunctional mitochondria [3839]. These results suggest that UA reversed the IS-induced reduction in the mtDNA content by maintaining mitochondrial fusion functions. Mitochondrial biogenesis promotes an increase in mitochondrial content to meet intracellular energy demands [40]. Downregulation of mitochondrial biogenesis leads to muscle atrophy and subsequent detrimental consequences for exercise capacity in patients with CKD [4142]. Mitochondrial biogenesis is activated by the expression of mitochondrial and nuclear genes that are regulated by transcription factors, such as Ppargc1a, Nrf1, Tfam, Sirt1, and Mef2c. In particular, Ppargc1a is a well-known pivotal regulator of mitochondrial biogenesis. In this study, the mRNA expression levels of Ppargc1a, Nrf1, and Tfam were decreased upon treatment with IS; however, this decrease was improved upon treatment with UA (Fig. 3D). Ppargc1a promotes the transcriptional activity of Tfam by binding to and co-activating Nrf1 on the Tfam promoter [43]. Tfam activates mtDNA replication and transcription, and the interaction between Tfam and mtDNA is involved in the regulation of mitochondrial biogenesis [44]. These results revealed that UA improved the IS-induced reduction in mitochondrial biogenesis and mtDNA content by regulating the expression of Ppargc1a, Nrf1, and Tfam. In addition, the expression of Ppargc1a is mediated by Sirt1 and Mef2c [45]. In this study, the mRNA expression levels of Sirt1 and Mef2c were decreased by treatment with IS; however, the decrease was improved upon treatment with UA (Fig. 3D). These results also indicate that UA ameliorates the IS-induced reduction of mitochondrial content in C2C12 cells by regulating the expression levels of the abovementioned genes.
A recent study reported that muscle mitochondrial contents were decreased in CKD mice, and this decrease was related to an increased concentration of the inflammatory cytokine IL-6 in blood [46]. White et al. [47] reported that IL-6 overexpression induced muscle atrophy in a mouse model of colon cancer, and this induction was associated with the upregulation of Fis1 expression and downregulation of Ppargc1a, Mfn1, and Mfn2 expression. In addition, administration of the IL-6 receptor antibody ameliorates the IL-6-induced decrease in mitochondrial content and expression of Fis1, Ppargc1a, Mfn1, and Mfn2 [47]. In this study, IL-6 mRNA expression and protein secretion were increased upon treatment with IS; however, this increase was improved after treatment with UA (Fig. 4A and B). In addition, STAT3 activity was significantly increased after treatment with IS; however, this increase was improved by treatment with UA (Fig. 4C). While IL-6 activates many signaling pathways, the activation of STAT3, which is immediately downstream of IL-6, has been associated with the loss of mitochondrial functions in muscles [48]. These results suggest that UA improved the decrease in mitochondrial content by suppressing the IS-induced increase in IL-6 secretion.
Normal differentiated cells maintain viability and function through ATP production, which relies on mitochondrial OXPHOS. Fatty acid is a central fueling substrate for ATP production in mitochondria. In this study, IS decreased the mRNA expression of representative FAO genes (Pdk4, Acadm, Cpt1b, and Cd36); however, this decrease was improved by treatment with UA (Fig. 5A). These results indicate that UA contributes to the maintenance of energy production capacity by suppressing the IS-induced decrease in the expression of FAO-related genes. In addition, IS decreased the mRNA expression of representative OXPHOS genes (Cycs and Atp5b) and intracellular ATP levels; however, this decrease was improved upon treatment with UA (Fig. 5B and C). Intracellular ATP levels increase with the progression of myoblast differentiation to meet increased energy demands [49]. These results suggested that UA maintains normal differentiation by suppressing the decrease in IS-induced ATP levels in C2C12 cells.
In summary, the current study revealed that UA improved the IS-induced impairment in mitochondrial biogenesis, and its mechanisms were associated with muscle differentiation, ATP levels, and IL-6 secretion. Therefore, this study indicated that UA could be a potential therapeutic candidate for treating CKD-induced muscle dysfunction.
Go to :
 XML Download
XML Download