

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide [1]. The occurrence of metastasis in CRC is considered a highly fatal course [2]. However, the theory of oligometastases suggest that a reduced number of metastases represent a transitional state between localized and diffuse disease, and local control of the metastases might lead to improved outcomes [3,4]. In fact, under favorable settings of oligometastases, median survival may surpass 5 years in patients with solitary lung or liver metastasis [5,6]. On the other hand, there are some patients who rapidly recurred after potentially curative metastasectomy [7]. To date, there are no established guidelines for the selection of patients who require aggressive treatments to maximize their long-term survival and those for whom aggressive treatments may be harmful.
As clinical risk factors, the local stage of primary tumor, synchronicity of metastases, tumor marker level, number and size of metastases, and disease-free interval were reported to be significantly associated with tumor recurrence after metastasectomy [8,9]. However, these risk factors are hardly used in the clinical practice [10]. Some observations support that oligometastatic prognosis in CRC mainly relies on biological drivers rather than on clinical risk factors. Despite constant progress in imaging modalities, systemic treatments, and surgical techniques, the proportion of patients cured after metastasectomy has not significantly improved over recent decades [7].
Several mutational molecular markers identified in CRC include microsatellite instability, BRAF, and KRAS/NRAS, which confer poorer outcomes [11]. However, most of these studies are based on primary cancers, and little is known regarding the molecular profile of metastasis and its relation to clinical outcomes in oligometastatic cancers [12]. Moreover, emerging evidence of both intra- and inter-tumor heterogeneities in several solid tumor types have raised concerns that the molecular profiling of primary tumors may not be representative of metastatic disease [13-15]. Intra-tumor heterogeneity refers to differences within a single tumor or between primary and metastatic tumors, and inter-tumor heterogeneity refers to differences between patients [16]. Further studies are needed because the tumor heterogeneity can confuse the application of biomarkers that may predict treatment response or prognosis.
In this study, we aimed to identify the concordant or discordant genomic profiles between primary and matched metastatic tumors from patients with CRC undergoing metastasectomy and we attempted to classify genomic groups according to genomic discrepancy patterns and identify candidate genomic patterns that could affect survival after metastasectomy.
Materials and Methods
1. Patients and tumor samples
Patients with advanced or metastatic (stage IV) CRC treated at Korea University Anam Hospital (Seoul, Korea) between September 2005 and August 2018 were screened for the study. The criterion for study inclusion was metastasectomy. There were no exclusions for tumor histology, medical comorbidities, previous treatments, prior therapy, or performance status. Genomic profiling was performed using the Ion AmpliSeq Cancer Hotspot Panel (ICP) with the Ion Torrent Proton system.
2. Tissue genomic DNA extraction
For each tumor, hematoxylin and eosin–stained sections were reviewed by a gastrointestinal pathologist. When the tumor tissues were identified, tumors were macrodissected to maximize tumor content. Tissue gDNA was extracted from formalin-fixed, paraffin-embedded (FFPE) tumor tissues using the QIAamp DNA FFPE Tissue Kit (Qiagen, Valencia, CA) according to the manufacturer’s instructions and eluted in a 50-μL volume. Purity of the extracted gDNA was assessed by 1% agarose gel electrophoresis, and DNA concentration was quantified on a Qubit 2.0 Fluorometer using the Agilent High Sensitivity DNA Kit (Agilent Technologies, Santa Clara, CA).
3. Next-generation sequencing
Overall, up to 10 ng of gDNA was extracted from FFPE samples and amplified using the Ion AmpliSeq Library Kit 2.0 (Life Technologies, Carlsbad, CA) over 20 cycles, with barcoding for each sample. The library concentration was evaluated using QuantStudio Real-Time PCR System (Thermo Fisher Scientific, Waltham, MA). Each diluted library (100 pM) was amplified by emulsion PCR using the One-Touch Instrument (Life Technologies) and was enriched by the OneTouch ES Instrument (Life Technologies) using the Ion PI Hi-Q OT2 200 kit following the manufacturer’s instructions. Finally, sequencing was performed on an Ion Proton instrument (Life Technologies) using an Ion PI Hi-Q Sequencing 200 Kit (Life Technologies). Barcoded samples were loaded onto an Ion PI Chip v3. Sequencing, read mapping, and variant calling were performed with Ion Torrent Suite v5.0.4.0. The ICP genes are shown in S1 Table.
4. Variant annotation
Data analysis was carried out using Torrent Suite Software V.5.0 (Life Technologies). After alignment to the hg19 human reference genome, the Variant Caller plug-in was applied. Ion Reporter suite (Life Technologies) was used to filter polymorphic variants. In addition, all nucleotide variations present at less than 1% variant frequency were masked. This value was useful for identifying variations present at a relatively low frequency compared to the value of 5% used in previous studies, as passenger mutations were detected and matched samples were filtered. All detected variants were manually reviewed with the Integrative Genomics Viewer (IGV V.2.1, Broad Institute, Cambridge, MA). Next, the presence of the variants in common databases was evaluated using 5000Exomes, dbSNP, Canonical RefSeq Transcripts, ClinVar, DGV, DrugBank, Gene Ontology, COSMIC, OMIM, Pfam, PhyloP Scores, RefGene Functional Canonical Transcripts Scores, RefSeq GeneModel, and Named Variants databases. Pathogenic variants were annotated as “likely-pathogenic” or “pathogenic” in the COSMIC and ClinVar databases (Fig. 1). Considering the functional significance of pathogenic variants, this study analyzed the non-synonymous variants expected to have deleterious effects. Targeted sequencing using the ICP panel generated approximately 100 Mb per sample with an average of 96.63% samples on target. Sequences of all samples achieved a mean depth of 3,900×.

5. Study design and definition
The present study identified the concordant or discordant genomic profiles by comparing genetic variants between primary and metastatic tumors and explored their clinical implications. We defined concordant variants as those present in both primary tumors and their matched metastatic tumors. Primary-specific variants were defined as existing only in primary tumors and metastasis-specific variants were defined as existing only in metastatic tumors. Using these genomic discrepancy patterns, we classified genomic groups related to survival. Overall survival (OS) was defined as the period from metastasectomy to death. Disease-free survival (DFS) was defined as the period from metastasectomy to recurrence and/or progression. Additional analyses examined the correlation between paired genetic mutations and metastatic sites and compared two paired metastatic tumors (M1 and M2).
6. Statistical analysis
All statistical analyses were performed using R ver. 3.1.2 software (https://www.r-project.org/) and SPSS ver. 22.0 (IBM Corp., Armonk, NY). Descriptive statistics were summarized as frequency distributions for categorical variables and as means for continuous variables. McNemar’s test was used to analyze the gene frequencies of primary and matched metastatic tumors. Principal component analysis (PCA) and partitioning around medoids (PAM) clustering algorithms were used to classify genomic groups. Survival rates were analyzed by the Kaplan-Meier method and log-rank test. Univariate and multivariate analysis were performed using a Cox proportional hazard model to identify prognostic factors. Nominal variables were compared using Fisher exact test. We used chi-square test and a machine learning algorithm with random forest plots to identify the correlation between genomic pattern and metastatic sites. A Mann-Whitney test was used for average comparison. We used the complete linkage method to reconstruct the phylogeny of multiple specimens from individual patients based on the presence or absence of total variants. p-values of < 0.05 were considered statistically significant.
Results
1. Patient characteristics and data set
To compare the genomic profiles of primary and metastatic CRC, we collected 335 surgical samples from 158 patients. The samples consisted of 158 pairs of primary (P) and matched metastatic tumors (M1), and an additional 19 pairs of metastatic tumors (M2), with each pair derived from the same patient. Based on 158 pairs of P and M1 tumors, the mean age at metastasectomy was 61 years and the group comprised 96 men (60.8%). The initial stages were as follows: six stage I (3.8%), 24 stage II (15.2%), 49 stage III (31.0%), and 79 stage IV (50.0%). The type of metastasis was defined as synchronous metastasis when the primary and metastatic tumor tissues were collected within 6 months (n=71, 44.9%) and metachronous metastasis when the difference was greater than 6 months (n=87, 55.1%). The most common sites of primary tumors were the rectum (n=80, 50.6%), left colon (n=54, 34.2%), and right colon (n=24, 15.2%). The most common sites of metastases were the liver (n=95, 60.1%), followed by the lungs (n=56, 35.4%), and others (n=7, 4.4%). Detailed patient characteristics are provided in Table 1.
Table 1.
Main characteristics of the patients
![]()
Screening of a sequence of 50 genes from the 316 samples (158 pairs of P and M1 tumors) revealed 744 types of variants (Fig. 1). Most of these variants have been previously described in the COSMIC or ClinVar databases (412 of 744, 55.4%). Among them, 310 of 412 (75.2%) were pathogenic variants or likely pathogenic variants. Of these 310, we analyzed 282 (91.0%) non-synonymous variants, excluding coding silent, in-frame indels, and intronic variants, which were expected to have a deleterious effect. The present study defined these as deleterious non-synonymous variants. The number of types of deleterious non-synonymous variants per patient, varying from 0 to 27, is presented in S2 Table.
2. Genomic profiling between primary and matched metastatic tumors
The baseline genomic profiling between primary (P) and matched metastatic tumors (M1) are shown in Fig. 2. In the frequencies of deleterious non-synonymous variants between primary and metastatic tumors, TP53, KRAS, APC, and PIK3CA displayed the largest numbers of variants in the primary CRC and their metastases (Fig. 2A). There was no overall statistical difference in the frequency of mutations between primary and metastatic tumors. However, the frequency of mutations was numerically higher in primary tumors than in metastatic tumors in KRAS, APC, KIT, FBXW7, SMAD4, PTEN, and CDKN2A. In particular, APC mutations showed the greatest difference between primary and metastatic tumors; however, the difference was not statistically significant (32.3% vs. 27.2%, McNemar’s test p=0.057). The overall mutation frequencies were consistent with the expected mutation frequencies for CRC samples reported by the Cancer Genome Atlas (TCGA). However, APC mutations were observed less frequently in our study compared to TCGA (primary, 32.3%; metastasis, 27.2% vs. TCGA 73.2%).
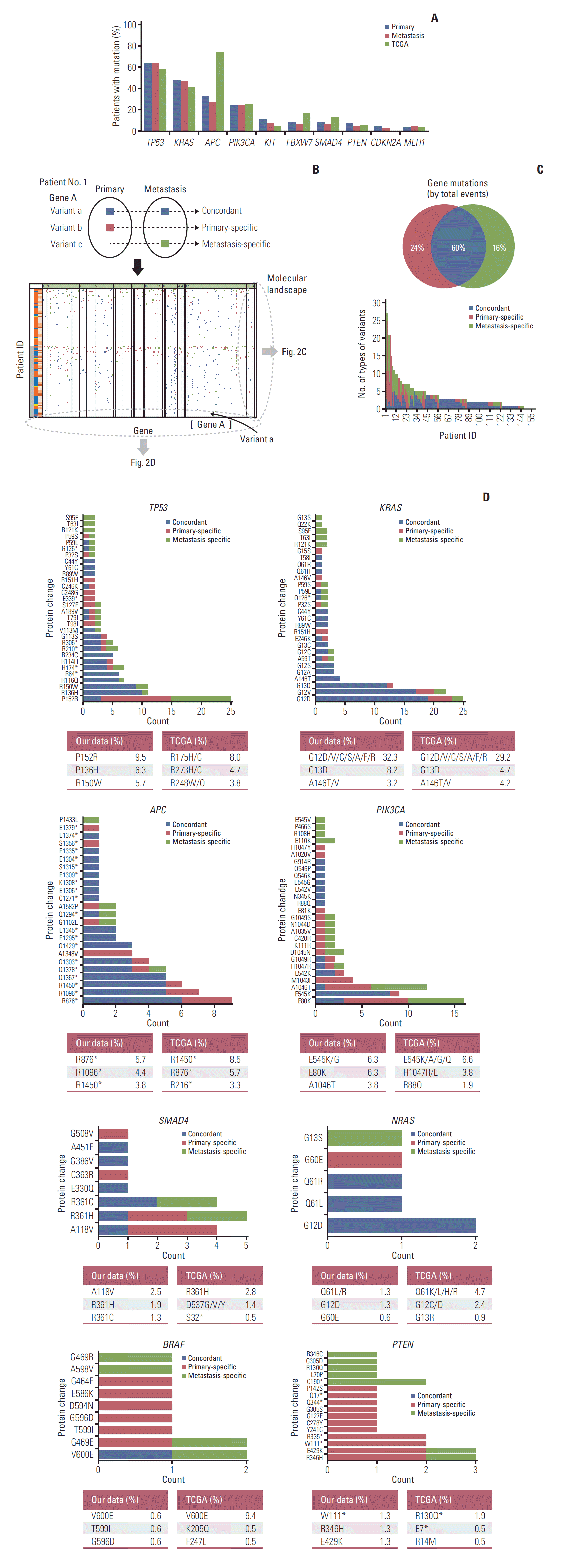
Fig. 2.
Genomic profiling between primary tumors (P) and matched metastases (M1). (A) Frequencies of deleterious non-synonymous variants between primary and metastatic tumors compared with the Cancer Genome Atlas (TCGA) data. (B) The schematic concept of concordant, primary-specific, and metastasis-specific variants and schematic illustration of mutational landscape. (C) Comparison of the percentage of mutations identified as primary-specific, metastasis-specific, or concordant between primary and matched metastases. Twelve patients who did not have deleterious non-synonymous variants were excluded from the comparison. The distribution of number of types of variants per patient is shown at the bottom. (D) Frequencies based on the different protein changes of key genes and comparison with TCGA data. X, variant count (number of variants); Y, each position. Different protein changes represent different variant positions. TP53 shows only the top 30 variant positions. The highest frequencies of the top three variant’s positions compared to the TCGA data are shown at the bottom of each graph. The data were calculated based on 158 primary tissue samples. TCGA data were obtained from 212 tissue samples.
![]()
To identify intra-tumor heterogeneity, we identified concordance and discordance of mutations between primary and matched metastatic tissues. A schematic diagram is shown in Fig. 2B. We classified genetic variants into concordant, primary-specific, and metastasis-specific variants. We obtained the overall mutational landscape of genetic variants for 158 patients (S3 Fig.). From the mutational landscape, we identified the distribution of variants according to each patient (Fig. 2C) and each gene (Fig. 2D).
Among all deleterious non-synonymous variants, an average of 60% of events were concordant and an average of 40% of events were discordant between the primary and matched metastasis: 24% for primary-specific variants and 16% for metastasis-specific variants (Fig. 2C). The total number of concordant variants was 538 (269 concordant variants were doubled because of their presence in both primary and metastatic tumors) (S2 Table), whereas the total number of discordant variants was 359. The average number of concordant variants per patient was 3.4 and discordant variants per patient was 2.3. The discordant variants were caused more frequently by primary-specific variants (215/359, 60%), compared with the metastasis-specific variants (144/359, 40%). The distribution of the number of types of variants per patient is shown at the bottom of Fig. 2C.
Fig. 2D depicts the frequencies based on the protein changes of key genes that were observed in patients with CRC. Different protein changes represented different variant positions. The portion of concordant mutations was large in TP53, KRAS, APC, and NRAS, whereas the portion of discordant mutations was large in PIK3CA, SMAD4, BRAF, and PTEN. The frequencies of the protein changes in primary tumors were compared with those of TCGA data. KRAS, PIK3CA, NRAS, and BRAF showed similar protein change frequency order as the TCGA data. The most common protein changes were KRAS G12D, PIK3CA E545K/G, NRAS Q61L/R, and BRAF V600E. TP53, APC, SMAD4, and PTEN differed from TCGA data. The most common protein changes were TP53 P152R, APC R876*, SMAD4 A118V, and PTEN W111* in our data.
3. Genomic grouping and survival analysis
As shown in the genomic profiling, varied information for concordant, primary-specific, and metastasis-specific variants was observed. PCA was used to convert a set of observations of possibly correlated variables, including number of types of variants and variant positions, into a set of values of linearly uncorrelated variables (principal components, PC). Three distinct PCs (PC1, PC2, and PC3) were used for PAM clustering algorithm. Using the silhouette method, the optimal number of genomic groups to classify the patients was found to be three. The three groups were classified as shown in Fig. 3A and S4 Table. Detailed comparison of clinical characteristics between genomic groups was shown in S5 Table.
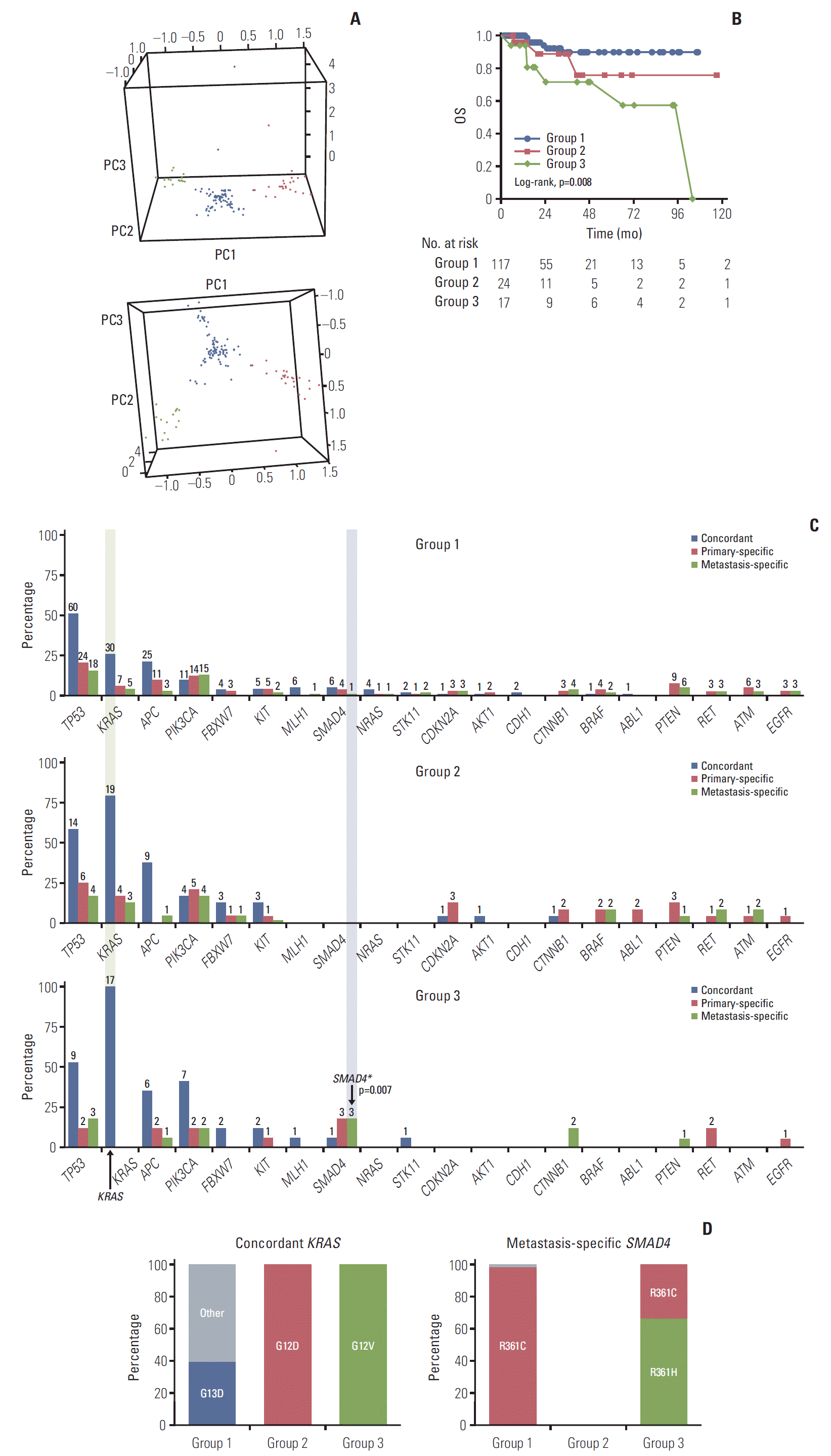
Fig. 3.
Genomic grouping and survival analysis. (A) Three genomic groups were determined using clustering with PC1, PC2, and PC3; group 1 (117, 74.0%, blue), group 2 (24, 15.2%, red), and group 3 (17, 10.8%, green). (B) Overall survival (OS) data. The OS of group 3 was significantly shorter than that of group 1. (C) Genetic frequency between groups and the concordant, primary-specific, and metastasis-specific genes. The frequency of the top 20 concordant (blue), primary-specific (red), and metastasis-specific (green) genes. X denotes each gene, Y denotes percentage of patients with the mutation in each group, and labels denote the number of patients. (D) The proportion of variants of concordant KRAS and metastasis-specific SMAD4 between genomic groups.
![]()
OS differed significantly between the genomic groups (overall log-rank p=0.008) (Fig. 3B). The OS of group 3 was significantly shorter than that of group 1 (p=0.002). This was maintained even after adjusting for other variables, including age at metastasectomy, sex, initial stage, type of metastasis, primary site, metastatic site, and sum of diameter for metastatic lesions at metastasectomy using a multivariate Cox regression model (overall p=0.009) (Table 2). In subgroup analysis by genomic group, OS was significantly shorter for patients in group 3 than for those in group 1 (hazard ratio, 6.98; 95% confidence interval [CI], 2.01 to 24.20; p=0.002). However, there was no significant difference in the DFS (overall log-rank p=0.252) (not shown in the Fig. 3B).
Table 2.
Multivariate analysis of variables associated with overall survival
![]()
To identify the characteristics of each group, the frequency and specific protein changes of major genes were confirmed. In the frequency analysis, group 1 with good survival showed the lowest frequency for overall concordant, primary-specific, and metastasis-specific genes (Fig. 3C). Group 2 with unclassified survival showed an overall median frequency between that of groups 1 and 3. However, with regard to concordant TP53, group 2 showed the highest frequency. Group 3 with poor survival showed the highest frequency of concordant KRAS (100%) and concordant PIK3CA (41.2%) mutations, and five metastasis-specific genes were higher than those in the other groups: TP53, APC, SMAD4, CTNNB1, and PTEN.
To investigate the characteristics of group 3 with poor survival, the proportion of protein changes in the characteristic genes of group 3 was compared with other groups (Fig. 3D). For concordant KRAS mutations, which most exclusively divided the group in the frequency analysis, group 1 with good prognosis had the highest proportion of G13D, whereas group 3 with poor prognosis had 100% of G12V. Conversely, group 2 had 100% of G12D and was clearly distinct from group 3. The proportions of other major genes, such as concordant TP53, APC, and PIK3CA, were also compared; however, they did not show unique protein changes in group 1 or 3 and they were commonly distributed (S6 Fig.). For the metastasis-specific genes, six genes were identified in group 3: TP53, APC, PIK3CA, SMAD4, CTNNB1, and PTEN. Among the six metastasis-specific genes in group 3, SMAD4 was found to be a significant metastasis-specific gene that differentiated group 3 and the other groups (Fisher exact test p= 0.007). And for metastasis-specific SMAD4, group 3 with poor prognosis had the highest proportion of R361H (Fig. 3D). The proportions of additional metastasis-specific genes are shown in S6 Fig. Although there were some unique proportions of protein changes in group 1 and 3, there was no statistical basis to distinguish group 3 from the others.
4. Correlation between paired genes and metastatic sites
Whether specific paired genes affected the metastatic sites was assessed using the chi-squared test and random forest model. We determined the correlation between paired genes (concordant, primary-specific, and metastasis-specific) and overall metastatic sites (liver only, lungs only, both liver and lungs, and others) in the patients during their overall follow-up period (Fig. 4). For the concordant genes, KRAS was significantly correlated with metastases beyond the liver, such as the lungs and other sites, i.e., more disseminated metastases in the chi-squared test (p=0.014). In the random forest model, KRAS, APC, TP53, KIT, and SMAD4 were identified as the top five genes, in that order, with the greatest influence when the conditional effect due to the interaction of each gene was considered in the metastatic sites. In the primary-specific genes, BRAFdisplayed a significant correlation with lung metastasis in the chi-squared test (p=0.002). In the random forest model, TP53, BRAF, RET, KRAS, and PTEN were identified as the five most influential genes. Concerning the metastasis-specific genes, TP53 and PTEN displayed a significant correlation with both liver and lung metastases (p=0.004 and p=0.016, respectively). In the random forest model, TP53, PTEN, KRAS, PIK3CA, and ATM were identified as the five most influential genes. Detailed percentage of overall metastatic sites according to the mutated paired genes were shown in S7 Fig.
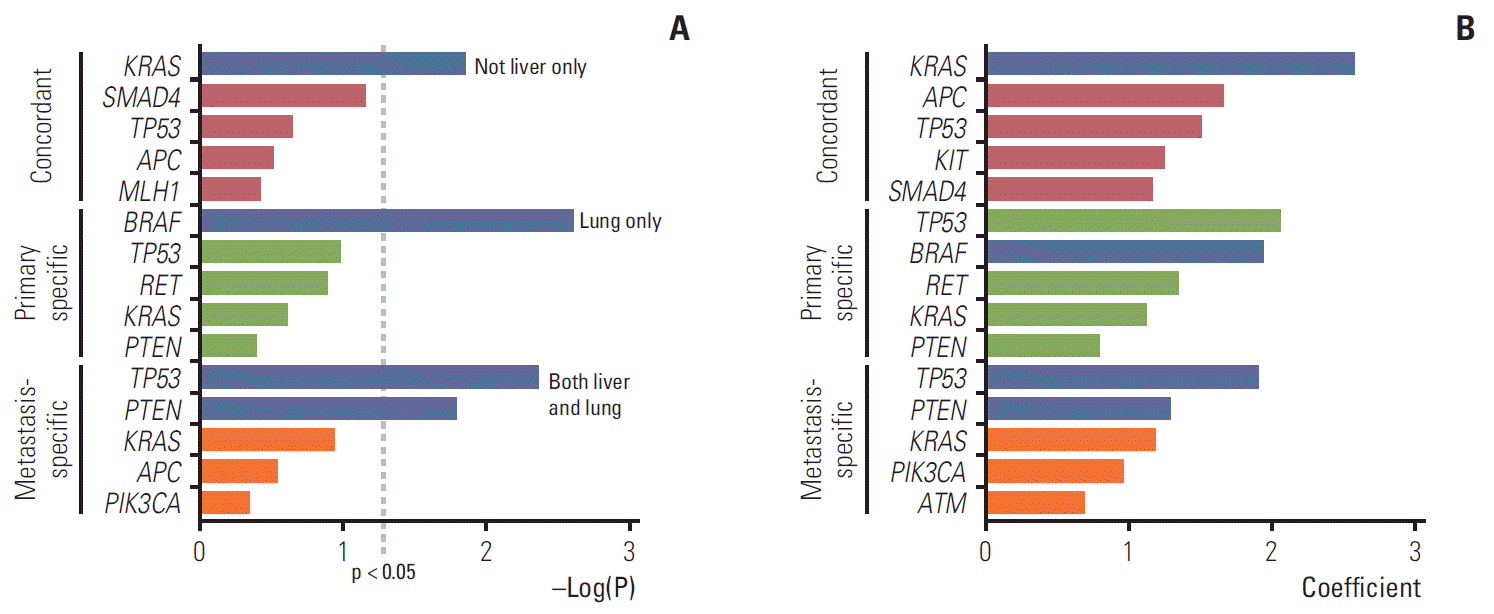
Fig. 4.
Correlation between paired genes (concordant, primary-specific, and metastasis-specific) and metastatic sites (liver only, lung only, both liver and lung, and others). (A) Chi-square test. Blue indicates p < 0.05. (B) Random forest model. In the random forest and lasso regression model, the top five genes with a high coefficient were selected. Blue indicates genes that were significant in the chi-squared test.
![]()
5. Comparative analysis with two paired metastatic tumors (M1 and M2)
Two paired metastatic tumors (M1 and M2) in 19 patients were further analyzed. M2 was tissue obtained through metastatic resection at the same time or after M1. Most metastatic tumors of M2 were from the lungs (n=12, 63.2%). However, liver (n=5, 26.3%), brain (n=1), and peritoneum (n=1) metastases were also represented. To identify changes in intra-tumor heterogeneity through additional M2 tissues, we applied a linkage method to construct phylogenetic trees according to the distance between P, M1, and M2 (S8 Fig.). According to the relationship between P and M1, synchronous and metachronous metastasis were classified.
In patients with synchronous metastases, the genetic origin of M2 followed P rather than M1 (primary-driven progression; 9/10, 90%) (Fig. 5A). On the other hand, in patients with metachronous metastases, the genetic origin of M2 followed M1 sequentially (sequential; 3/9, 33.3%) (Fig. 5A). There were four cases where the genetic origin could not be identified because P, M1, and M2 were identical in all hot spots in metachronous metastasis. The extent of heterogeneity was determined according to the different number of total variants between P and M1 or M2. The average number of total variants was 24 for synchronous metastasis and 10 for metachronous metastasis (Mann-Whitney test p=0.053) (Fig. 5B).
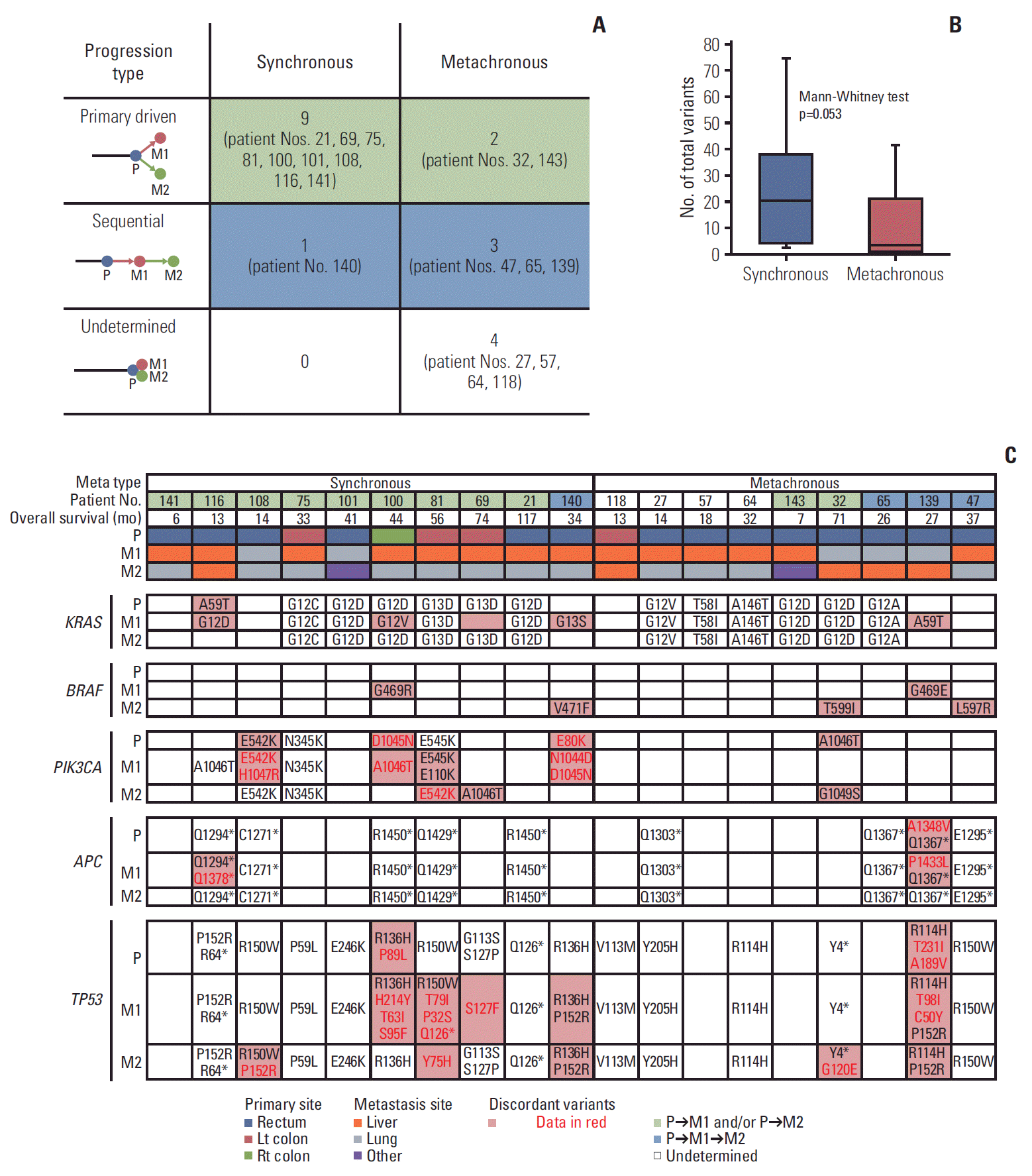
Fig. 5.
Mutations between primary and metastatic tumors for a subgroup of 19 patients with M1 and M2 tumors. (A) Progression type according to synchronous and metachronous metastasis. (B) Heterogeneity according to synchronous and metachronous metastasis. (C) Variant changes of key genes in colorectal cancer between P, M1, and M2.
![]()
KRAS, BRAF, PIK3CA, APC, and TP53 were found to be the key genes based on their variants (Fig. 5C). KRAS variants appeared consistently in the lungs, whereas KRAS variants were not consistently present in the liver. BRAF mutations coexisted with KRAS mutations in the same tumor tissue in three patients: BRAF G469R/KRAS G12V, BRAF G469E/KRAS A59T, and BRAF T599I/KRAS G12D. PIK3CA mutations appeared more in synchronous metastasis. APC and TP53 mutations showed relatively consistent mutations up to M2. More than two-hit mutations of TP53 were frequently observed.
Discussion
Advances in next-generation sequencing technology in recent decades make that knowledge of the biological drivers of cancer will lead to personalized cancer treatment [13,17]. Indeed, the accumulation of molecular alterations of these key driver genes plays a crucial role in the tumorigenesis and progression of CRC. However, it remains controversial, as previous studies usually rely on a limited sample of cancer tissue that cannot represent heterogeneity between and within patients [18]. This study is an integrated analysis of intra-tumor and inter-tumor heterogeneity. We found the overall intra-tumor heterogeneity by comparing primary and metastatic tumors, and we yielded the genomic groups based on the inter-tumor heterogeneity between patients. We also obtained some notable correlations between paired genes and metastatic sites by considering the tumor heterogeneity.
First, in demographics, there was a lower prevalence of APC genes in the Korean population compared with TCGA data. The frequency of APC mutations between our data and TCGA data may vary owing to the definition of the mutations. TCGA data included truncating mutations (putative driver) and missense mutations (unknown significance). However, since our data only included pathogenic or likely pathogenic variants classified in the COSMIC or ClinVar databases, the frequencies of APC gene were reduced by 32.3% and 27.2% for primary and metastatic tumors. In our data, when we extend our mutation definition as total variants, the frequencies of APC gene were up to 81% and 72.8% for primary and metastatic tumors, respectively. The majority of APC gene mutations are deletions or insertions or nonsense mutations, which result in the formation of a truncated APC protein [19]. However, in our data, only 36.8% of total variants have been classified as either pathogenic or likely pathogenic variants in the clinical significance value of ClinVar or COSMIC. Consistent with our data, Inra et al. [20] reported that the frequencies of pathogenic APC variants in 8,676 personal and/or family history of colorectal polyps and/or CRC were 17.5% overall, 25.2% in Asians, and 15.5% in Caucasians. As more than 50% of mutations in APC gene have uncertain or unknown clinical significance, further studies are needed to evaluate the clinical significance value of APC mutation and to accumulate more updated data.
Next, we showed there was an average of 40% genomic discrepancy between primary and matched metastases. More discordant variants were caused by variant presence in the primary CRC tumors and absence in the metastases. The ratio of primary-specific variants to metastasis-specific variants was 3:2. Indeed, primary CRCs could include a large cohort of passenger mutations, of which approximately 71.4% occur in the matching metastases. In previous studies, Brannon et al. have reported a 100% concordance for KRAS, NRAS, and BRAF between primary and metastatic CRCs [21]. However, in their study, the primary tumor and metastasis were simultaneously resectioned in 75% of patients. In addition, 43% of patients were chemotherapy-naive prior to resection. In our study, there is a time interval between primary and metastatic tumor resections (55.1% of patients exhibit metachronous metastasis; mean time interval between resections of metachronous metastasis is 24.9 months), and 86.1% of our patients were administered chemotherapy after primary resection. Chemotherapy after primary resection can cause genetic changes through cancer cell necrosis. For these reasons, discordances might be more in this study compared with that in the study by Brannon et al. Furthermore, we identified the frequency and position of each variant of key genes for concordant, primary-specific, and metastasis-specific variants. The portion of concordant variants was large in TP53, KRAS, APC, and NRAS, which are early mutations in CRC. In contrast, the portion of discordant mutations was large in PIK3CA, SMAD4, BRAF, and PTEN, which are known as later mutations. This can be explained through heterogeneous clonal evolution [22].
In terms of clonal evolution, additional intra-tumor heterogeneity was identified in 19 patients with two serial metastatic tumors. Interestingly, in synchronous metastasis, primary and metastatic tumors simultaneously showed different biology, as in the Big Bang model [23]. However, when M2 occurs, the genetic origin of M1 is followed by P, instead of sequential M1, which suggests the high effect of primary tumors on synchronous metastasis; it appears born-to-bebad. On the other hand, in metachronous metastasis, biology was similar between primary and metastatic tumors, and it developed in a more sequential way according to the accumulation of mutations. In CRC, the driver gene of the primary tumor plays a pivotal role in most cases. However, in some patients, changes in metastasis-specific genes can cause clonal evolution in a different direction than primary cancer, as demonstrated by patients 140 and 139, i.e., both showed simultaneous metastasis. TP53 P152R played a pivotal role in these two patients (S8 Fig.). Further research is needed on metastasis-specific mutations and their mechanisms, which play a pivotal role in clone evolution.
We used PCA and clustering to generate genomic groups based on inter-tumor heterogeneity that reflects intra-tumor heterogeneity. The genomic groups classified based on genomic discrepancy patterns showed significant differences in OS. Genomic groups remained significant even after correcting for other clinical risk factors, thus, suggesting that biological factors strongly influence the clinical outcome. Concordant KRAS G12V and metastasis-specific SMAD4 were identified as characteristics of the poor OS group (group 3). KRAS mutations occur early during the progression from colorectal adenoma to malignant carcinoma. Of note, the Ras-Raf-MEK-ERK and phosphoinositide 3-kinase/AKT pathways are strongly interconnected. Activation of this network by muta-tion deregulates the survival, mobility, and proliferation of cells [24]. In a recent study, specific mutations in KRAS codon 12 were associated with shorter OS in patients with advanced and recurrent CRC, especially in G12V [25]. This result is consistent with our results. Moreover, we found that no newly acquired metastasis-specific KRAS mutations were observed in group 3. Groups 1 and 2 accounted for more than 20% of metastasis-specific KRAS G12V (S6 Fig.). This suggests that concordant KRAS G12V had a more significant effect on survival than newly acquired KRAS G12V in metastasis.
As the pivotal factor of the transforming growth factor β pathway, which regulates tumorigenesis and tumor progression, SMAD4 mutations were reported to be present in 2.1%-20.0% of CRC [26]. In a meta-analysis, Huang et al. [18] reported that patients with CRC who had a KRAS mutation (combined odds ratio [OR], 1.29; 95% CI, 1.13 to 1.47) or SMAD4 mutation (combined OR, 2.04; 95% CI, 1.41 to 2.95) were at a higher risk of distant metastasis. No significant association was found between mutations of APC or PIK3CA and CRC metastasis [18]. Considering that the occurrence of distant metastasis is associated with poor prognosis, this is consistent with our results. However, while the results of previous studies were obtained from an OR of each gene, our study considered several genes concurrently. Considering these results, patients who had KRAS G12V mutation in the primary tissue at the time of diagnosis should confirm the genetic changes or mutations present in the metastatic tissue when they have to decide the treatment directions or to predict the prognosis. However, even in patients who did not have this mutation, it is necessary to confirm those of metastatic tissues before determining the treatment directions as it helps in identifying mutations with poor prognosis, such as SMAD4.
In other aspects of primary and metastasis research, Joung et al. [27] classified oligo-clones and multi-clones based on the number of subclones in primary and metastatic tumors using a hierarchical Bayesian clustering model [27]. There was a significant difference in the DFS between the oligoclone and multi-clone groups, but a difference in OS was not proven. This result differed from our findings, because they quantified heterogeneity that did not reflect the types of genetic mutations. We assessed all the components, including the discrepancy pattern between primary and metastatic tumors and types of genetic variants. Overall, these findings suggest that the degree of heterogeneity is related to DFS, but it is the genetic mutations that affect the OS.
To the best of our knowledge, the present study is the first to document a preference for metastatic sites based on paired genes. These findings correlated well with our survival data. Concordant KRAS that occupied a high proportion of group 3 tended to disseminate and were not confined to one organ. When a random forest model was applied, the metastatic sites were predicted by the conditions of several genes frequently found in CRC. Datta et al. [28] reported that metastatic CRC tumors harboring co-occurring RAS/BRAF and TP53 alterations were significantly more likely to involve extrahepatic metastatic sites compared with tumors that were RAS/BRAF-altered or TP53-altered only. This analysis considered the synergistic effects of the three genes, and the results for key genes were similar to our findings. However, our study considered the conditional effects of 50 genes that can be detected. The results showed extrahepatic metastasis in concordant KRAS and metastasis-specific TP53 and PTEN. Primary-specific BRAF was more likely to be associated with lung metastasis. Fidler [29] insisted that clone survival was dependent on the surrounding environment, such as the liver and lungs, by the seed-soil hypothesis. Further research is required on the mechanisms of clonal selection according to metastatic sites and their interactions with other genetic mutations.
The present study had several limitations. First, we included only deleterious non-synonymous variants. In recent studies, synonymous variants with deleterious effects have been reported [30]. For instance, synonymous variants may create a new splicing site or obliterate an existing one, thus, turning an exonic sequence into an intron, or vice versa, which results in the production of a different polypeptide. However, in our study, most of the synonymous variants had a high frequency of silent variants and few had no exact function or effect. Second, the criteria for oligometastasis was not clear. Although, we included patients with oligometastases occurring in resectable CRC, the study population may be a heterogeneous group depending on whether it is a debulking operation or staged operation. However, this heterogeneity was corrected for by the clinical factors, including metastatic types and sum of the diameter of metastasis lesions at the time of metastasectomy. Third, the present study is a model for the current data set. This model may have an overfitting problem and is only significant in this enrolled patient group; therefore, validation with larger sample sizes is required. Fourth, compared with the average number of deleterious non-synonymous variants per patient (4.0), nine patients had more than 10 deleterious non-synonymous variants. Five out of nine patients were confirmed low microsatellite instability/microsatellite stable and the microsatellite instability status of four could not be identified. One case out of nine corresponded to mucinous carcinoma. However, seven of the nine patients were in group 1 and two patients were in group 2. Although it was not possible to determine the overall MSI status, we could rule out the possibility that group 3 with poor prognosis was biased to patients with a high number of variants.
In conclusion, driver gene mutations were mostly concordant; however, discordant or metastasis-specific mutations were present. Clinically, the concordant driver genetic changes, especially in KRAS G12V mutations, with additional metastasis-specific variants, especially in SMAD4 mutations, can predict poor prognosis in patients with CRC. These findings suggest that we must consider the biology of the metastatic tumor compared with the primary tumor, as this can help guide treatment and predict the survival of patients with oligometastatic CRCs.
 XML Download
XML Download