

 PDF
PDF Citation
Citation Print
Print


Introduction
Jab1 (c-Jun activation domain binding protein-1, also known as CSN5 [COP9 signalosome subunit 5]) is a co-activator of c-Jun, a transcription factor involved in numerous cellular processes including cell proliferation and carcinogenesis [1]. Jab1 is encoded by the COPS5 gene and enhances the transcriptional function of c-Jun by binding to c-Jun. An earlier study proved that Jab1 binds to and promotes the translocation of p27 from the nucleus to the cytoplasm, decreasing p27 expression in the cell by 26S proteasomemediated degradation [2,3]. In addition, ectopic expression of Jab1 in murine fibroblasts partially overcame p27-mediated G1 cell cycle arrest. Jab1 also promotes the nuclear export and cytoplasmic degradation of p53 in coordination with HDM2 [4,5].
Jab1 overexpression is frequently observed and inversely correlated with p27 expression in a variety of cancers including breast cancer [6], ovarian cancer [7], hepatocellular carcinoma [8], esophageal cancer [9], and laryngeal cancer [10]. Several studies have also shown that Jab1 expression is associated with the prognosis of patients with various cancers [7-10]. Therefore, Jab1 could be a potential therapeutic target in cancer [11].
Biliary tract cancer (BTC) is relatively rare in Europe and North America, but frequent in Asia, including Korea and Latin America [12]. Recent development of targeted agents has significantly improved the prognosis of patients with various cancers. However, there is still no clinically validated therapeutic target in BTC. For BTC patients, only gemcitabine combined with cisplatin has been established as a standard chemotherapy [13]. Therefore, there is a huge need for the development of therapeutic targets for patients with BTC.
Although the role of Jab1 has been established in other solid tumors, no studies have examined the expression of Jab1 in BTC or the potential therapeutic implication of Jab1 expression in BTC. In this study, we examined the expression and function of Jab1 in human BTC cell lines, and evaluated the therapeutic potential of Jab1 inhibition in BTC using in vitro and in vivo models with the aim to examine the possibility of Jab1 inhibition for the treatment of BTC patients.
Materials and Methods
1. Human BTC cell lines
A total of eight human BTC cell lines were used in this study. SNU245, SNU308, SNU478, SNU869, SNU1079, and SNU-1196 cell lines were obtained from the Korean Cell Line Bank (Seoul, Korea) [14]. HuCCT-1 and TFK-1 cell lines were purchased from RIKEN BioResource Center (Ibaraki, Japan). Each cell line was authenticated using the AmpFLSTR Identifiler PCR Amplification Kit (catalog No. 4322288; Applied Biosystems, Foster City, CA) by the Korean Cell Line Bank on March 8, 2016. The 3530xL DNA Analyzer (Applied Biosystems) and the GeneMapper v5 (Applied Biosystems) were used for DNA fingerprinting analysis. All the cell lines were maintained in RPMI-1640 media containing 10 μg/mL gentamicin and 10% fetal bovine serum (FBS; Welgene Inc., Gyeongsan, Korea) in a humidified atmosphere containing 5% CO2 at 37°C.
2. Jab1 knockdown by siRNA and shRNA and chemotherapeutic agent
We used siRNA for transient silencing of the COPS5 gene that encodes Jab1 and shRNA for stable knockdown of the COPS5 gene. Jab1-specific or control (nonspecific) siRNAs were obtained from Genolution Pharmaceuticals, Inc. (Seoul, Korea). The sequence of the Jab1-specific siRNA was 5′-GCTCAGAGTATCGATGAAA-3′. The sequence of the control siRNA was 5′-AATTCTCCGAACGTGTCACG-3′. All cells were transfected with siRNAs at a final concentration of 50 nM using Lipofectamine 2000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. After daily transfection for 2 days, the cells were harvested and subjected to western blot analysis.
Jab1-specific or control shRNAs and lentiviral particle gene silencers were purchased from Santa Cruz Biotechnology (Dallas, TX); the shRNAs and gene silencers are a pool of concentrated, transduction-ready, viral particles containing three target-specific constructs that encode 19-26 nt shRNAs designed to knock down gene expression. The shRNAs were transduced to the cell lines according to the manufacturer’s instructions. Stable clones that expressed the shRNA were selected using puromycin dihydrochloride (Santa Cruz Biotechnology). Western blot analysis was used to confirm the target gene expression in these stable clones.
Cisplatin was purchased from JW Pharmaceutical Co. (Seoul, Korea).
3. Western blot analysis
Cells were lysed in RIPA buffer containing protease inhibitors on ice for 15 minutes. Protein samples were obtained by centrifugation at 13,000 rpm for 20 minutes. Equal amounts of proteins were separated on 10% sodium dodecyl sulfate polyacrylamide gels and transferred onto nitrocellulose membranes. The membranes were incubated at 4°C overnight with primary antibodies. Primary antibodies against the following molecules were purchased from Cell Signaling Technology (Beverly, MA): p53, RAD51, PTEN, AKT, phosphorylated AKT (Ser473), Src, and phosphorylated Src (Tyr416). Following antibodies were purchased from Santa Cruz Biotechnology: cyclin D1, cyclin E, cyclin A, Jab1, γH2AX, and p27. Anti-α-tubulin and anti-β-actin antibodies were purchased from Sigma-Aldrich (St. Louis, MO). Antibody binding was detected using an enhanced chemiluminescence system according to the manufacturer’s protocol (Amersham Biosciences, Piscataway, NJ). Anti-mouse and rabbit secondary antibodies were purchased from Thermo Scientific Inc. (Waltham, MA). The data was quantified by ImageJ software (National Institute of Health, Bethesda, MD) and normalized by α-tubulin or β-actin as an internal control.
4. Cell growth inhibition assays
For 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assays, cells transfected with Jab1-specific or control siRNA or shRNA were seeded in 96-well plates: 2,000 cells per well for the SNU478 cell line and 4,000 cells per well for the HuCCT-1 cell line. MTT dye (Sigma-Aldrich) was added to each well and cells were incubated for 4 hours at 37°C. The MTT solution was removed and dimethyl sulfoxide was carefully added. Cell viability was calculated by measuring the absorbance at 540 nm with a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA). Six wells were used for each experimental condition.
For colony formation assays, SNU478 cells and HuCCT-1 cells were seeded in six-well plates at a density of 500 cells and 3,000 cells, respectively. The cells were then treated with Jab1-specific or control siRNAs. After 10 days, the cell colonies were stained with 0.1% Coomassie Blue solution (Sigma-Aldrich), and counted using a Gel Doc (Bio-Rad, Hercules, CA). The data presented are representative of three independent experiments.
5. Cell cycle analysis
Cells transfected with Jab1-specific or control siRNAs were harvested, fixed with cold 70% ethanol, and stored at −20°C. The fixed cells were harvested by centrifugation, and incubated with 20 mg/mL RNase (Invitrogen) at 37°C for 10 minutes. Cells were then stained with 20 μg/mL propidium iodide (Sigma-Aldrich). The DNA content of 10,000 cells per each experimental group was analyzed using a FACS Calibur flow cytometer (BD Biosciences, Franklin Lakes, NJ). Three independent experiments were performed for each condition.
6. Wound healing assays
Cells transfected with Jab1-specific or control siRNA or shRNA were grown as monolayers in six-well culture plates. Confluent monolayers were gently scratched with a sterile 200-μL pipette tip. The plates were then washed with phosphate-buffered saline (PBS). After 24 hours, cell movement back into the area of the scratch was recorded by light microscopy. The area of each wound was quantified by ImageJ software (National Institute of Health). Ten wounds were examined at once for each experimental condition. The data presented are representative of three independent experiments.
7. Transwell migration assays
The transwell migration assays were performed using transwell plates (Corning Costar, Cambridge, MA) which were 6.5 mm in diameter with 8 μm pore filters. SNU478 and HuCCT-1 cells were treated with Jab1-specific or control siRNA once daily for two days. Then, a cell suspension containing 1×105 cells/mL in 200 μL RPMI-1640 media and 0.1% FBS were added to top chambers. In addition, lower chambers contained 500 μL RPMI-1640 media with 10% FBS. After incubation for 24 hours, cells in the top chambers were removed and then 4% paraformaldehyde was treated for 20 minutes. The invading cells were stained with 0.1% crystal violet for 10 minutes, and then examined using light microscope. The crystal violet was dissolved in 300 μL of 33% acetic acid. The solution was then quantified by measuring the absorbance at 573 nm with a VersaMax microplate reader (Molecular Devices). The data presented are representative of two independent experiments.
8. Comet assays
Jab1-specific or control siRNAs were transfected into SNU-478 and HuCCT-1 cell lines daily for 2 days. These cell lines were treated with 2.5 μM or 5 μM cisplatin for 48 hours. The cells were then trypsinized and subjected to an alkaline comet assay using the Trevigen Comet Assay Kit (Trevigen Inc., Gaithersburg, MD) following the manufacturer’s protocol. Tail moments and tail intensity were measured with Comet Assay IV software (Perceptive Instruments Ltd., Bury St. Edmunds, UK). Three independent experiments were performed for each condition.
9. Real-time quantitative polymerase chain reaction
RNA was extracted using TRI Reagent (Molecular Research Center, Inc., Cincinnati, OH) according to the manufacturer’s instructions. Then, 2 μg of total RNA was used to reverse transcribe cDNA with random hexamers and ImProm-II Reverse Transcriptase (Promega, Madison, WI). Real-time quantitative polymerase chain reaction (PCR) was performed by the StepOnePlus System (Applied Biosystems) using Premix Ex Taq (Takara Bio Inc., Shiga, Japan) with SYBR Green I (Molecular Probes, Eugene, OR) to validate gene expression. All data were normalized to actin cDNA levels. The experiments were performed in duplicate.
10. Cycloheximide chase assays
Cycloheximide chase assays were used to quantify the half-life of proteins. SNU478 and HuCCT-1 cells were transfected using Jab1-specific or control siRNAs. Transfected cells were seeded in plates and cycloheximide 20 μg/mL was added to the plates. Cells were collected at indicated time points for western blot analysis.
11. In vivo study
Female BALB/c athymic nude mice aged 4-6 weeks were purchased from Central Lab Animal Inc. (Seoul, Korea). The mice were subcutaneously injected in the right flank with 5×107 Jab1-specific or control shRNA-transfected HuCCT-1 cells in 200 μL of PBS. Four mice for each group were used for tumor formation. Two weeks after HuCCT-1 cell injection, tumor formation was evaluated. After implantation of the tumor cells, the size of the tumors was measured every other day using calipers for 4 weeks. The tumor volume was calculated using the following formula: (width)2×(height)/2. At 4 weeks, the tumors were excised and stored in liquid nitrogen until further analysis by immunohistochemical staining and western blot.
12. Immunohistochemistry
Sections of 4-μm thickness from paraffin-embedded tumor tissues were deparaffinized and dehydrated. Anti-Jab1 antibody (Santa Cruz Biotechnology) at a dilution of 1:2,000 and anti–Ki-67 antibody (GeneTex, Inc., Irvine, CA) at a dilution of 1:100 were used to detect proliferating cells in the tumor samples. Terminal deoxynucleotidyl transferase–mediated dUTP nick end labeling (TUNEL) assays were performed to evaluate apoptosis in the tumor samples using an ApopTag In situ Apoptosis Detection Kit (EMD Millipore, Billerica, MA), in accordance with the manufacturer's protocol.
Results
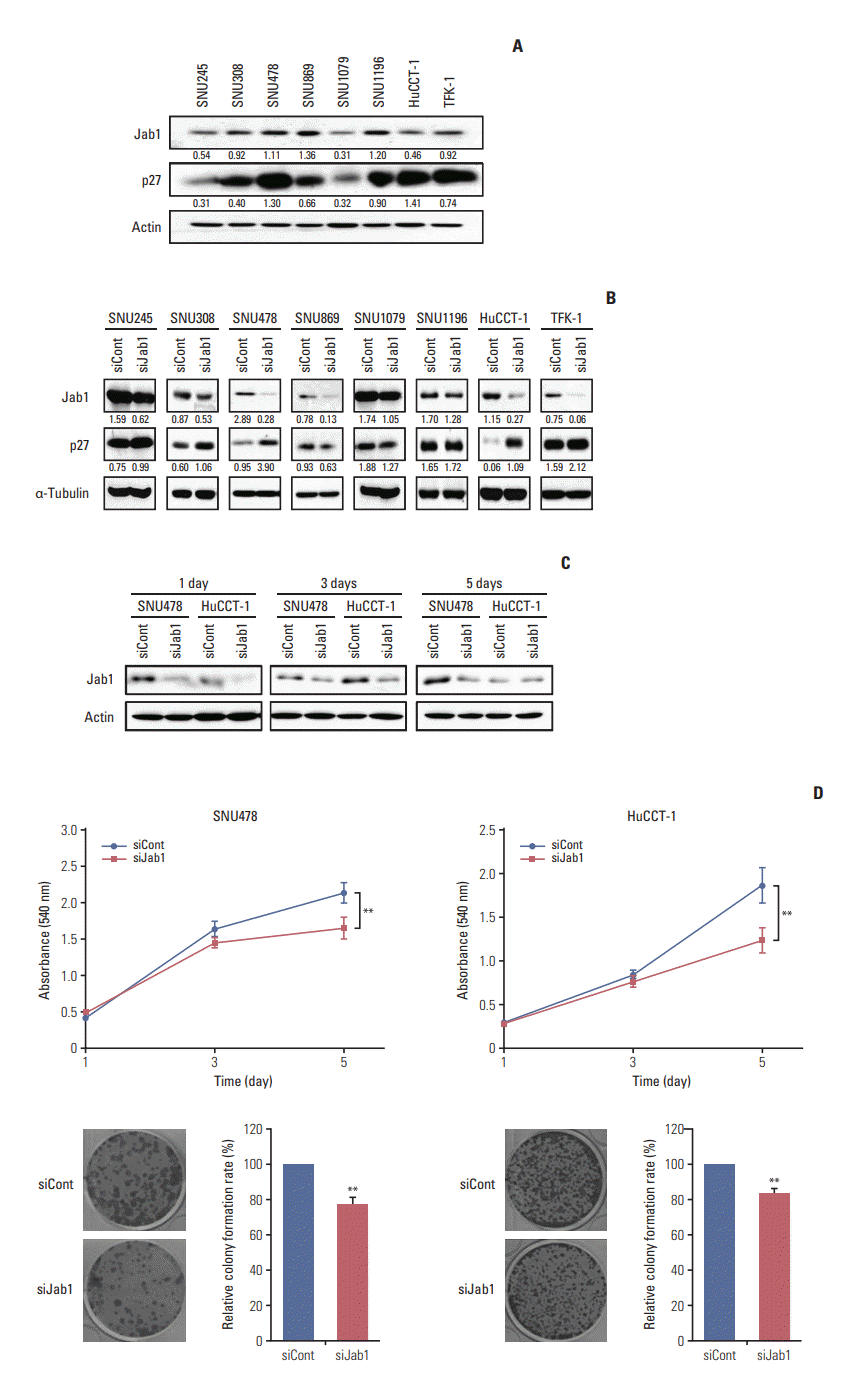
1. Jab1 silencing by siRNA increases p27 expression in BTC cells
We first examined the expression of Jab1 in eight BTC cell lines. All BTC cell lines showed a various range of Jab1 and p27 expressions (Fig. 1A). Among the cell lines examined, SNU478, SNU869, and SNU1196 cells had relatively higher Jab1 expression levels. Of these three, SNU478 and SNU1196 cell lines showed relatively higher basal p27 expression as well. In contrast, SNU245, SNU1079, and HuCCT-1 cell lines had relatively lower basal Jab1 expression levels.
Previous studies demonstrated that Jab1 decreases p27 expression levels and is inversely correlated with p27 in several cancers [6-10]. Our results also showed that Jab1 silencing by siRNA increased p27 expression in six of eight BTC cell lines (Fig. 1B). In particular, in SNU478 and HuCCT-1 cell lines, Jab1-specific siRNA strongly knocked down Jab1 expression and resulted in increased p27 expression. Realtime quantitative PCR experiments reproduced the findings of the western blot analysis (S1 Fig.). We hypothesized that Jab1 inhibition increases p27 expression in some BTC cell lines, resulting in anti-proliferative activity. However, the amplitude of corresponding p27 induction by Jab1 silencing was not associated with basal Jab1 expression levels of the cell lines we tested. We thus selected SNU478 and HuCCT-1 cell lines for further in vitro and in vivo experiments.
Knock-down effects of Jab1-specific siRNA were maintained at 5 days in SNU478 cells, while diminished at 5 days in HuCCT-1 cells (Fig. 1C). To evaluate the effect of Jab1 on cell proliferation, we performed MTT and colony formation assays. Our results showed that Jab1 silencing significantly retarded the growth of SNU478 and HuCCT-1 cells (Fig. 1D), indicating that Jab1 silencing inhibited the proliferation of BTC cells.
2. The Cancer Genome Atlas data analysis validates the cell line findings
To validate this finding in BTC samples, we analyzed The Cancer Genome Atlas provisional data of BTC using cbioportal. org as of August 6, 2017. Among 36 BTC patients whose OncoPrint data was available, Jab1 (COPS5 gene) mRNA up-regulation defined by Z-score more than 2.0 was observed in five patients (13.9%) (S2 Fig.). Notably, one of five up-regulated patients had Jab1 gene amplification. In addition, the reverse phase protein array (RPPA) data demonstrated that Jab1 protein was overexpressed in two patients (5.6%). These data suggest that a substantial portion of patients has Jab1 mRNA or protein overexpression or gene amplification.
The RPPA analysis indicated that basal Jab1 protein expression levels were positively correlated with basal p27 (CDKN1B) protein levels with a Pearson coefficient of 0.78 (S3 Fig.). In addition, patients with Jab1 alteration tended to have better overall survival than those without Jab1 alteration (p=0.052) (S4 Fig.).
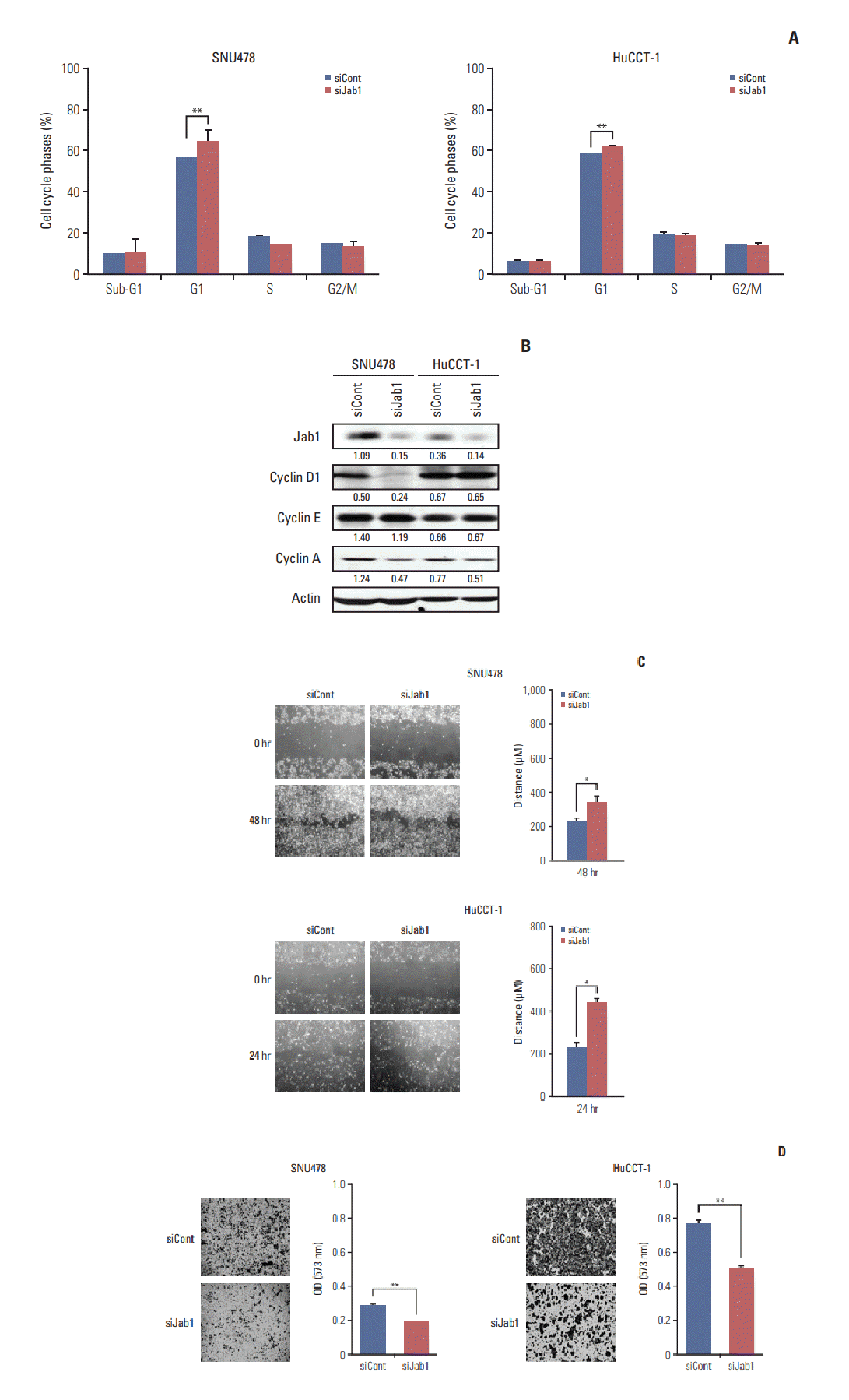
3. Jab1 silencing induces G1 arrest and inhibits migration of BTC cells
We next examined the growth inhibitory effect of Jab1 silencing in BTC cells in more detail. Cell cycle analysis showed that Jab1 silencing significantly increased the fraction of cells in G1 phase in SNU478 and HuCCT-1 cell lines (Fig. 2A). In addition, Jab1 silencing decreased cyclin D1 expression in SNU478 cells and cyclin A expression in the two cell lines (Fig. 2B).
We next evaluated the effect of Jab1 silencing on the cell migration capacity of BTC cells. In wound healing assays, Jab1 silencing significantly delayed the migration of SNU478 and HuCCT-1 cells (Fig. 2C). Transwell migration assays were then performed to evaluate the effects of Jab1 knockdown on cancer cell invasion (Fig. 2D). Jab1 knock-down significantly decreased the invasive activity of SNU478 and HuCCT-1 cells.
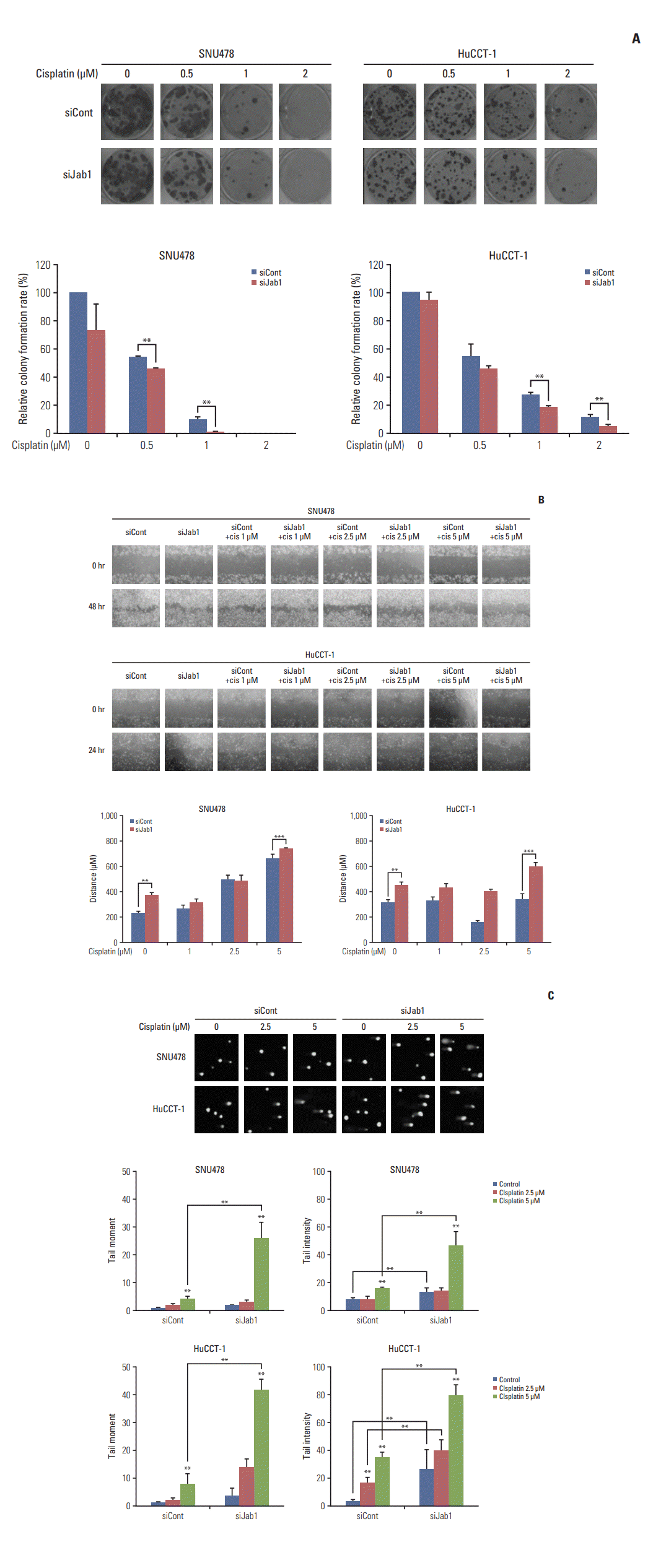
4. Jab1 silencing sensitizes BTC cells to cisplatin
To investigate the effects of Jab1 silencing on cisplatin treatment in cells, we performed colony formation assays. Jab1-specific or control siRNAs were transfected into SNU-478 and HuCCT-1 cell lines daily for 2 days. Cells were then treated with cisplatin at various concentrations (0, 0.5, 1.0, and 2.0 μM) for 10 days and colony formation was monitored. Results showed that Jab1 silencing significantly potentiated the anti-proliferative effects of cisplatin at various concentrations in the two cell lines (Fig. 3A). Furthermore, in wound healing assays, Jab1 silencing significantly enhanced the anti-migratory effects of cisplatin (Fig. 3B). In comet assays, both tail moments and intensities were significantly increased in proportion to the increased concentration of cisplatin, and the tail intensities were significantly increased by Jab1 silencing alone without cisplatin (Fig. 3C). Notably, Jab1 silencing significantly increased both the tail moments and intensities when combined with cisplatin at 2.5 and 5.0 μM.
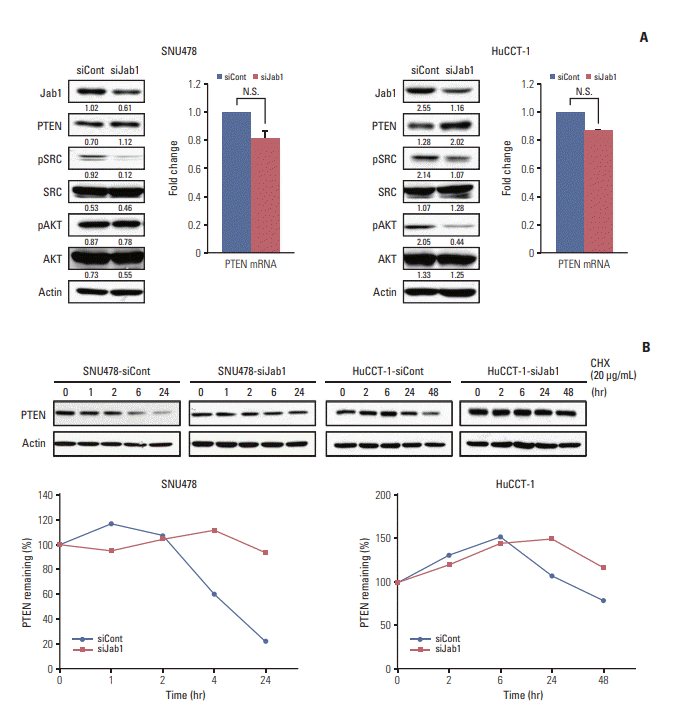
5. Jab1 knock-down inhibits Src phosphorylation via PTEN stabilization
We investigated the effects of Jab1 on the Src signaling pathway since Src is one of key molecular targets in BTC in our recent studies [15,16]. Jab1 silencing decreased phosphorylation of Src Tyr416 in SNU478 cells and AKT Ser473 and Src Tyr416 in HuCCT-1 cells (Fig. 4A). In a previous study of HER2-positive breast cancer, increased PTEN expression deactivated Src phosphorylation and vice versa [17]. Therefore, we examined the change of PTEN protein expression levels after Jab1 knock-down. Interestingly, Jab1 silencing increased PTEN expression levels in SNU478 and HuCCT-1 cells, without significant increase of PTENmRNA expression levels. In cycloheximide chase assays, Jab1 silencing increased the half-life of PTEN protein compared with the control in SNU478 and HuCCT-1 cells (Fig. 4B).
6. Stable knockdown of Jab1 by shRNA increases p27 expression and decreases proliferative and migratory activity
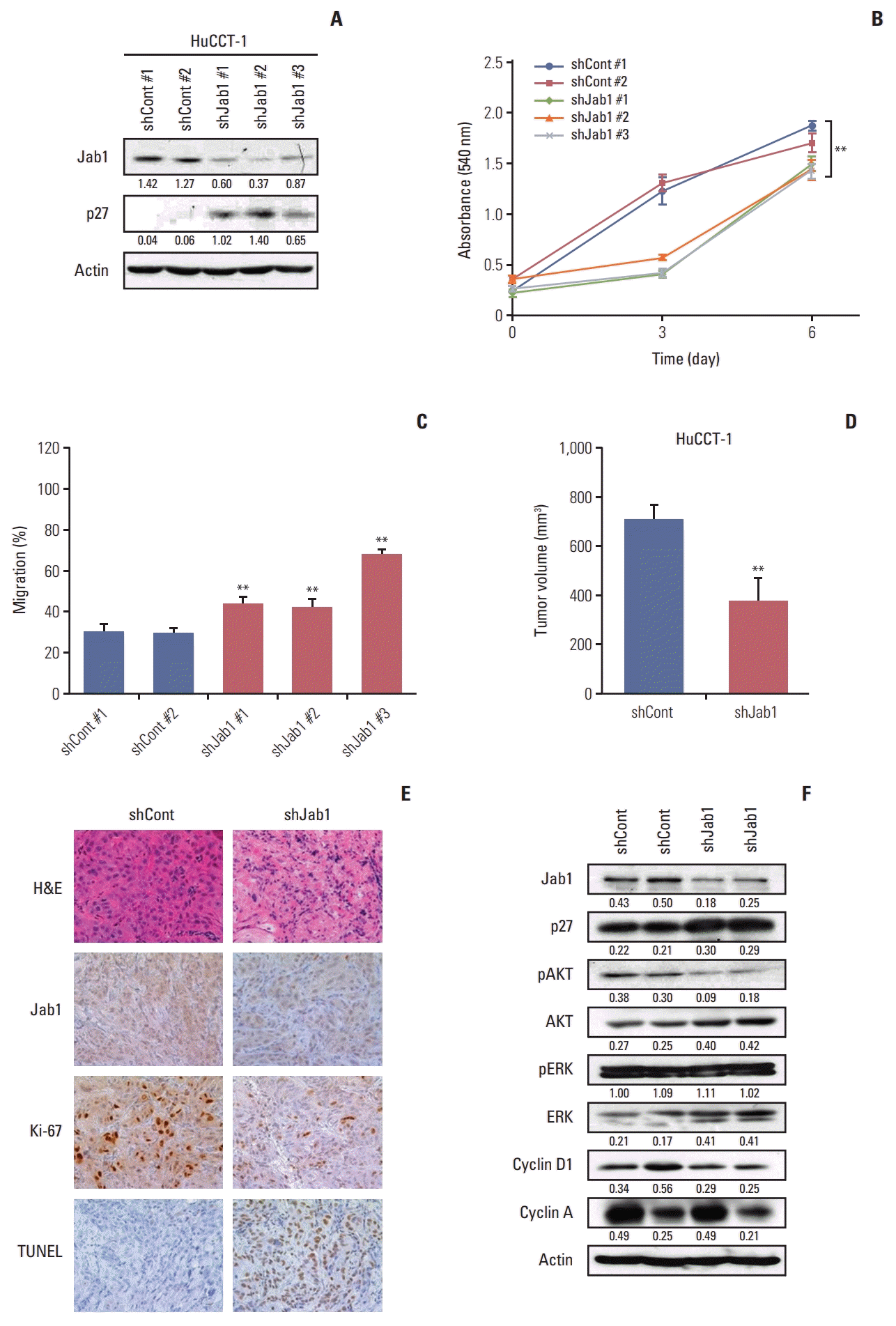
We next examined the in vivo effects of Jab1 silencing in a HuCCT-1 mouse xenograft model. We first developed HuCCT-1 cells that were stably knocked down for Jab1 by shRNA (S5 Fig.). In line with the results of Jab1 silencing by siRNA, stable knockdown of Jab1 by shRNA increased p27 expression levels (Fig. 5A) and significantly decreased proliferative (Fig. 5B) and migratory activities (Fig. 5C) compared with the control in HuCCT-1 cells.
In the HuCCT-1 mouse xenograft model, mice injected with HuCCT-1 cell stably knocked down for Jab1 by shRNA showed significantly decreased tumor volume in vivo (Fig. 5D). In immunohistochemical staining, tumor xenografts with silenced Jab1 expression showed decreased Ki-67 expression and increased TUNEL expression (Fig. 5E). Tumors with silenced Jab1 expression showed increased p27 expression, decreased AKT phosphorylation, and decreased cyclin D1 expression in vivo (Fig. 5F).
Discussion
In this study, Jab1 expression levels were relatively higher in most of the examined BTC cell lines. Moreover, Jab1 knockdown also increased p27 expression in BTC cells, which is in line with the results of previous studies in other cancers [6-10]. Jab1 knockdown produced anti-proliferative and anti-migratory effects in BTC cells by increasing DNA damage, stabilizing PTEN, and leading to G1 cell cycle arrest. In addition, Jab1 inhibition potentiated the anti-proliferative effects of cisplatin. Our in vitro experimental data was reproduced in an in vivo mouse model using HuCCT-1 cell lines with stable Jab1 knockdown by shRNA.
To the best of our knowledge, this is the first report on the role of Jab1 in BTC. In a previous study, Jab1 deficiency in mice resulted in early embryonic death and increased radiation-induced apoptosis by spontaneous DNA damage and homologous recombination defects [18]. In addition, Jab1 depletion inhibited proliferation of pancreatic [19], colorectal [20], gastric [21], and nasopharyngeal cancer cell lines [22] in vitro. In line with these results, our data suggest that Jab1 inhibition could also be a novel therapeutic target in BTC.
In our data, Jab1 knock-down increased p27 expression and induced G1 cell cycle arrest, which corresponds to previous data [2]. A previous study showed that p27 abrogation increased both basal and serum-stimulated transcription of cyclin D1 and A genes [23]. In our data, Jab1 silencing decreased cyclin D1 expression in SNU478 cells and cyclin A expression in SNU478 and HuCCT-1 cells. In the HuCCT-1 in vivo model, Jab1 knockdown decreased cyclin D1 expression more prominently than cyclin A. Therefore, our data of BTC suggest that G1 cell cycle arrest induced by Jab1 silencing may be attributed to the increased p27 expression resulting in decreased cyclin D1 and A expression.
Interestingly, in nasopharyngeal carcinoma, Jab1 silencing potentiated the anti-proliferative effects of DNA damaging agents including cisplatin, ionizing radiation, and ultraviolet radiation in vitro [22,24]. In our data, cisplatin significantly increased the anti-proliferative and anti-migratory effects of Jab1 silencing in BTC cells. Moreover, when combined with cisplatin, Jab1 silencing significantly increased both the tail moments and intensities in comet assays. Notably, the tail intensities were significantly increased by Jab1 silencing even without cisplatin treatment. These results suggest that Jab1 depletion spontaneously induces DNA damage and increases the anti-proliferative and anti-migratory effects of DNA damaging agents.
In colorectal cancer cell lines, Jab1 silencing decreased p38 and AKT phosphorylation [25]. Inversely, in breast cancer cell lines, HER2 transcriptionally increased Jab1 expression via AKT/β-catenin pathways, and HER2 inhibition decreased Jab1 expression [26,27]. Therefore, these data suggest that Jab1 and AKT are associated with each other. In our data in HuCCT-1 cells, Jab1 silencing decreased phosphorylation of AKT both in vitro and in vivo. In contrast, in SNU 478 cells, Jab1 silencing did not influence AKT phosphorylation. In addition, in HER2-positive breast cancer, increased PTEN expression deactivated Src phosphorylation and vice versa [17]. In our data, increased PTEN expression by Jab1 silencing deactivated Src phosphorylation in both SNU478 and HuCCT-1 cell lines, which is in line with the findings in HER2-positive breast cancer. Our results suggest that the anti-proliferative effects of Jab1 silencing in BTC may be mediated by both the phosphoinositide 3-kinase/AKT pathway and Src pathway.
Previous studies of Jab1 in several cancers have been performed using in vitromodels of transient knockdown of Jab1 by siRNA. No study has performed in vivo experiments using stable knockdown of Jab1 by shRNA in human cancer cells. In our study, we established a mouse model of stable Jab1 knockdown by shRNA using the HuCCT-1 cell line. Our in vivo data confirmed the findings of our in vitro experiments in BTC. Stable knockdown of Jab1 resulted in anti-proliferative effects compared with the control group. In addition, Jab1 depletion decreased Ki-67 expression and AKT phosphorylation and increased TUNEL and p27 expression in vivo.
Our in vitro and in vivo experimental data suggest that Jab1 silencing alone or in combination with cisplatin has a promising anti-tumor activity in BTC. Therefore, Jab1 could be a potential therapeutic target in patients with BTC, which is worthy of further pre-clinical and clinical investigations.
 XML Download
XML Download