

 PDF
PDF Citation
Citation Print
Print


Introduction
Receptor tyrosine kinases (RTKs) and their ligand have been found to be genetically altered and overexpressed in various human cancers and therefore considered as therapeutic targets for cancer [1]. The mesenchymal-epithelial transition factor (MET) and its ligand, hepatocyte growth factor, are required for organ regeneration and tissue damage repair, which resemble the pathological process of tumor invasion [2]. Accordingly, aberrant activation of MET signaling cascades including multiple downstream effector pathways such as signal transducer and activator of transcription 3 (STAT3), rat sarcoma (RAS)/mitogen-activated protein kinase (MAPK), and phosphoinositide-3-kinase (PI3K)/AKT, occurs in many types of cancer; receptor crosstalk with other RTKs also has been observed [3]. MET can be inappropriately activated via mutations, amplification and/or overexpression [4-6].
Lung cancer is the leading cause of cancer-related death worldwide. Genetic alteration of MET has been detected in non-small cell lung cancer (NSCLC) and MET amplification has been reported in epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI)-naïve NSCLC patients, with a prevalence of 1.4% to 21% [7-10]. In NSCLC, amplification of MET results in constitutive kinase activity in the absence of its ligand; overexpression acts as an oncogenic driver and activator of downstream signaling pathways such as the PI3K/AKT pathway [5,11].
MET presents an attractive therapeutic target for cancers including NSCLC and amplification of MET is an excellent predictive marker of sensitivity to MET-TKIs [6,9,12,13]. Capmatinib (INC280, Novartis) is a highly potent and selective small molecule inhibitor of MET. The selectivity of capmatinib is > 10,000-fold for MET in human kinase assays [14]. In addition, capmatinib demonstrated potent inhibition of cell growth and MET-dependent survival signaling activity in MET-dependent cell lines and patient tumor [6,15]. Although a dramatic response to capmatinib was observed in MET-amplified NSCLC [15], acquired resistance to capmatinib is inevitable. Therefore, in vitro NSCLC cell line models are useful to identify the molecular mechanisms of resistance to capmatinib and establish strategies to overcome it.
In this study, we have established MET-amplified NSCLC cell lines that showed acquired resistance to capmatinib and evaluated its resistance mechanisms. We demonstrated that capmatinib-resistant NSCLC cells were dependent on alternative pathway activation. Despite acquired resistance, the combined inhibition of EGFR or PIK3CA dramatically suppresses cell proliferation and downstream signals of capmatinib-resistant NSCLC cell lines, which suggested that this combination could be an effective therapeutic strategy against MET-TKI resistance in NSCLC patients.
Materials and Methods
1. Cell lines and drugs
The EBC-1 cell line, which is an NSCLC cell line that harbors a MET amplification, was purchased from the JCRB Cell Bank (Osaka, Japan). Capmatinib-resistant EBC-1 cell lines (EBC-CR1, -CR2, and -CR3) were established via stepwise exposure to capmatinib at final concentrations of 1.5, 2.2, and 2.4 μmol/L, respectively and were not selected from one cell without cloning. EBC-CR1, -CR2, and -CR3 cells were maintained in 1 μmol/L of capmatinib over 2 months and selected due to diverse resistance mechanisms; these are referred to as capmatinib-resistant cell lines. All cell lines were incubated in RPMI1640 medium (Gibco, Carlsbad, CA) with 10% FBS, 2 mmol/L of L-glutamine (Gibco), and 1% penicillin/streptomycin (Gibco) in a 37˚C incubator. Capmatinib (MET inhibitor), crizotinib (anaplastic lymphoma kinase/MET inhibitor), afatinib (irreversible EGFR inhibitor), BYL719 (PI3Kα inhibitor), PD1730-74 (fibroblast growth factor receptor [FGFR] inhibitor), and BGJ398 (FGFR inhibitor) were purchased from Selleck Chemicals (Boston, MA). Capmatinib was prepared at 5 mmol/L and the other drugs were prepared as 10 mmol/L stock solutions in 100% dimethyl sulfoxide. Recombinant heparin-binding EGF-like growth factor (HBEGF) was purchased from Prospec (East Brunswick, NJ). All cell lines used in this study were authenticated by Short Tandem Repeat STR analysis using PowerPlex kit (Promega, Madison, WI).
2. Cell viability and apoptosis assays
To determine the sensitivity of the cell lines to these inhibitors in vitro, cells were seeded into 96-well microplates at 5,000 cells/well and incubated in medium for 24 hours prior to exposure to the drugs. The cells were incubated with serial dilutions of the inhibitors in a final volume of 200 μL/well for 72 hours. After the 72-hour incubation, Ez-cytox (Dogen, Seoul, Korea) was added to each well and the plates were incubated for 3 hours in a 37°C incubator. The optical density was measured at a test wavelength of 450 nm using an Eon Microplate Spectrophotometer (Biotech, Winooski, VT). The IC50 values were calculated by Sigma Plot 12 (Hulinks, Tokyo, Japan). Cells were seeded in 6 well plates at a density of 5×105 per wells for cell cycle and apoptosis analyses. Twenty-four hours later, the cells were treated with inhibitors for 72 hours. Both adherent and floating cells were harvested and stained with propidium iodide (1.0 mg/mL) and RNase A (10 mg/mL) (Sigma-Aldrich, St. Louis, MO). Quantification for the cell-cycle distribution and the sub-G1 population was performed by flow cytometric analysis using BD FACSCalibur (BD, East Rutherford, NJ) and ModFit LT verity software (Verity Software House Inc., Topsham, ME).
3. Western blot analysis and human phospho-RTK array
EBC-1 parental and EBC-CR1, -CR2, and -CR3 cell lines, grown to adequate densities in 6-well plates, were treated for 2 hours with capmatinib alone, 24 hours with the combination of capmatinib and afatinib, or 24 hours with BYL719. Rabbit monoclonal antibodies to detect phospho-MET (Tyr-1234/1235), phospho-EGFR (Tyr1068), phospho-STAT3 (Tyr705), phospho-AKT (Ser473), total MET, total EGFR, total AKT, total p44/42 MAPK, and glyceraldehyde 3-phosphate dehydrogenase, and mouse monoclonal antibody to detect total STAT3 were purchased from Cell Signaling Technology (Danvers, MA). Mouse anti-phospho-p44/42 MAPK (Thr-202/Tyr204) was purchased from Santa Cruz Biotechnology (Dallas, TX). Western blotting chemiluminescence reagents were purchased from GE Healthcare (Chicago, IL). All antibodies and reagents were used according to the manufacturers’ instructions.
Phospho-RTK was detected using the Human Phospho-RTK Array kit (R&D Systems, Minneapolis, MN). An LAS-3000 imaging system (Fuji Photo Film Co., Stanford, CT) was used for image analysis.
4. Co-immunoprecipitation
Cells were washed once with modified Dulbecco’s phosphate-buffered saline and lysed in buffer containing 0.025 M Tris, 0.15 M NaCl, 0.0001 M EDTA, 1% NP-40, and 5% glycerol (pH 7.4). Protein G/A agarose resin (Thermo Fisher Scientific, Waltham, MA) was incubated for 2 hours with rabbit monoclonal anti-MET antibody (Cell Signaling Technologies), anti-EGFR antibody (Cell Signaling Technologies) or rabbit IgG control (Invitrogen, Carlsbad, CA) as a negative control and cell lysate at 1 mg of protein was mixed with antibody-coupled resin (Cell Signaling Technologies) overnight at 4°C. Co-immunoprecipitation was performed using the Pierce Co-IP Kit (Thermo Fisher Scientific). Immunoprecipitates were washed, eluted, and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis. Western blot was performed as described above.
5. Genomic DNA analysis
Gene copy numbers were determined by quantitative polymerase chain reaction (PCR). Genomic DNA (gDNA) was extracted from cell lines using the Exgene Cell SV Kit (Geneall Biotechnology, Seoul, Korea). Quantitative PCR was used to determine the gene copy number as follows: gDNA was amplified with specific primers (S1 Table) and SYBR Green PCR Master Mix (Applied Biosystems, Waltham, MA) using the GeneAmp PCR System (Applied Biosystems); images were captured with the Gel Logic 200 Imaging System (Kodak, Rochester, NY). Gene copy numbers were extrapolated from an external standard curve generated by serial dilutions of human gDNA (Merck Millipore, Darmstadt, Germany). The PCR products were purified using the PCR Purification Kit (Invitrogen), and sequenced by bidirectional Sanger sequencing.
6. Quantitative reverse transcription PCR and 3′-mRNA sequencing
Total RNA was extracted from EBC-1 parental and resistant cell lines using the RNA Mini Kit (Invitrogen) and reverse transcribed using Superscript III Reverse Transcriptase (Invitrogen). The StepOnePlus Real-Time PCR System (Applied Biosystems) was used for amplification of cDNA and quantitative real-time reverse transcriptase polymerase chain reaction was performed using the SYBR Green PCR Master Mix (Applied Biosystems) with specific primers (S2 Table). Relative gene expression was calculated using the ΔΔCt (cycle threshold) method with β-actin as the reference gene. Sequencing of the 3′-mRNA was performed using Quantseq technology (Lexogen, Vienna, Austria). RNA was extracted from EBC-1 and resistant cell lines as described above. Hierarchical clustering was analyzed on log2-fold changes in gene expression using the MultiExperiment Viewer (Dana Farber Cancer Institute, Boston, MA). Normalized read counts were calculated as (log2) > 6, and genes with an adjusted p < 0.05 were considered significantly differentially expressed.
Results
1. Establishment of EBC-1 cells with acquired resistance to capmatinib
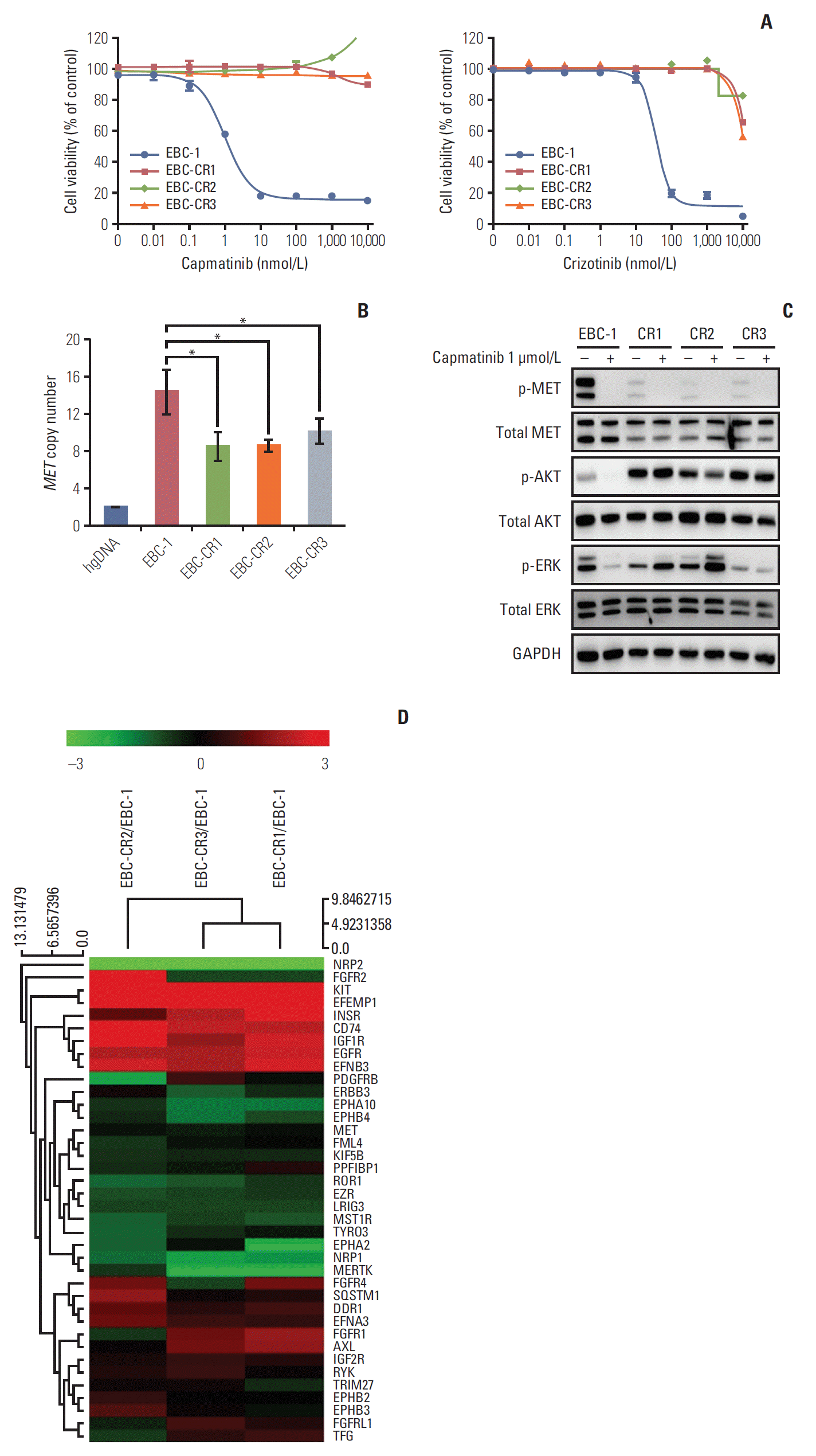
EBC-1 cells were derived from NSCLC cell lines harboring MET amplification and showing high sensitivity to MET-TKIs. To explore the molecular mechanisms of resistance to MET-TKIs, we used capmatinib, an MET inhibitor that is currently being studied in clinical trials. Using the EBC-1 cell line, we established total three cell lines resistant to capmatinib (EBC-CR1, -CR2, and -CR3) by stepwise exposure to capmatinib from 10 nM to final concentrations of 1.5, 2.2, and 2.4 μmol/L, respectively to observe various resistant mechanisms. The EBC-CR3 cell line was derived from the EBC-CR1 cell line by continuous exposure to a higher concentration of capmatinib during additional 3 months (S3A Fig.). First, we evaluated the sensitivity of the all three resistant cell lines to capmatinib by cell viability assay. The MET-TKIs were not toxic to the resistant cell lines (IC50, > 10 μmol/L on EBC-CR1–3 cells and 3.70±0.10 nmol/L on EBC-1 cells; and sub-G1, 18.15±3.31% on EBC-CR1, 8.37±4.61% on EBC-CR2, 12.17±3.6% on EBC-CR3, and 61.22±4.78% on EBC-1 cells) (Fig. 1A, S3B Fig.). Next, we confirmed the decrease of MET gene copy number by qPCR in resistant cell lines, which was accompanied by downregulation of MET phosphorylation (Fig. 1B). These results suggested that capmatinib strongly affected not only kinase activity but also MET copy number, which indicated its potent efficacy against MET-dependent tumors. Also, there were no additional MET-activating mutations identified by Sanger sequencing (data not shown).
In resistant cell lines, complete inhibition of MET phosphorylation by capmatinib demonstrated that there were no MET mutations that prevented the binding of capmatinib. Although complete inhibition of MET kinase was observed, phosphorylation of AKT and ERK1/2 were not completely inhibited in the resistant cell lines by a single treatment with capmatinib (Fig. 1C). These results suggest that ‘main’ survival pathways in resistant cells change from MET signal to other alternative kinase pathway.
2. Acquired resistant mechanisms to capmatinib associated with EGFR kinase pathway
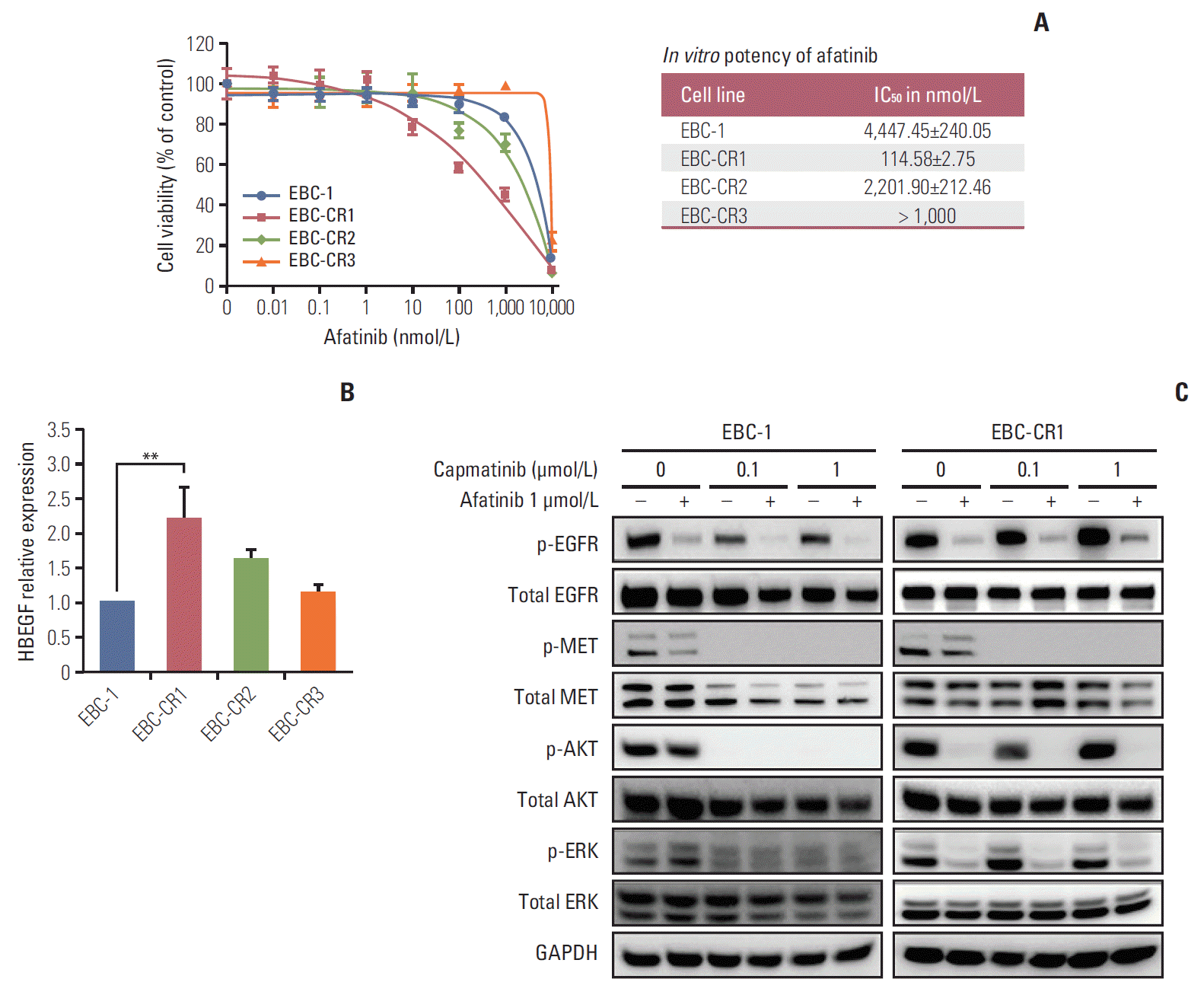
To identify alternative survival pathways of capmatinib resistance, EBC-1, EBC-CR1, -CR2, and -CR3 cells were screened for gene expression by 3′-mRNA sequencing. First, we analyzed expression of RTK-related genes by hierarchical clustering. RTK-related genes were stratified into several clusters and almost 20% of RTK-related genes were significantly upregulated in the resistant cell lines (Table 1, Fig. 1D). According to the results of 3' mRNA sequencing, MET mRNA expression was slightly decreased and EGFR mRNA expression was increased in EBC-CR1, -CR2, and -CR3 compared to the parental cell line (Table 1). We then confirmed increased EGFR protein phosphorylation compared to multiple RTKs in the resistant cell lines but observed no change in EGFR copy number (S1C and S3D Fig.). The mostly upregulated genes were KIT and EFEMP1 (Table 1, Fig. 1D). Although KIT mRNA was highly upregulated, KIT activation was not observed in phospho-RTK array (S3C Fig.). In contrast, EGFR signal was activated at both mRNA and protein levels. In addition, EFEMP1 that binds to EGFR can activate the downstream signals and eventually increase EGFR signal [16]. These results suggested that the EGFR signaling pathway might be an alternative pathway for MET. Although EGFR activation was increased in all resistant cell lines, cell viability assay to afatinib showed that only EBC-CR1 remained sensitive to afatinib alone (Fig. 2A). Interestingly, capmatinib inhibited EGFR kinase phosphorylation in EBC-1 cells. To determine the mechanism of EGFR pathway activation in the EBC-CR1 cell line, we tested the expression of multiple EGFR ligands (including epidermal growth factor [EGF], amphiregulin, epiregulin, transforming growth factor-α, and HBEGF) and observed that HBEGF expression was elevated in EBC-CR1 cells (Fig. 2B, S4A Fig.). To further explore the role of HBEGF in EGFR activation, we added exogenous HBEGF to the EBC-1 and EBC-CR1 cell lines and found that HBEGF was sufficient to rescue to MET inhibition in the MET-dependent NSCLC cell line, EBC-1 (S3E Fig.). In EBC-CR1, cultures with HBEGF were more resistant to EGFR inhibition than those without ligand (p < 0.01) (S3F Fig.). Unlike the parental cell line, a single treatment with afatinib effectively inhibited phosphorylation of EGFR, AKT, and ERK1/2 despite persistent MET kinase activation (Fig. 2C). As a result, EGFR activation was highly dependent on HBEGF as the main survival signal in EBC-CR1 cells.
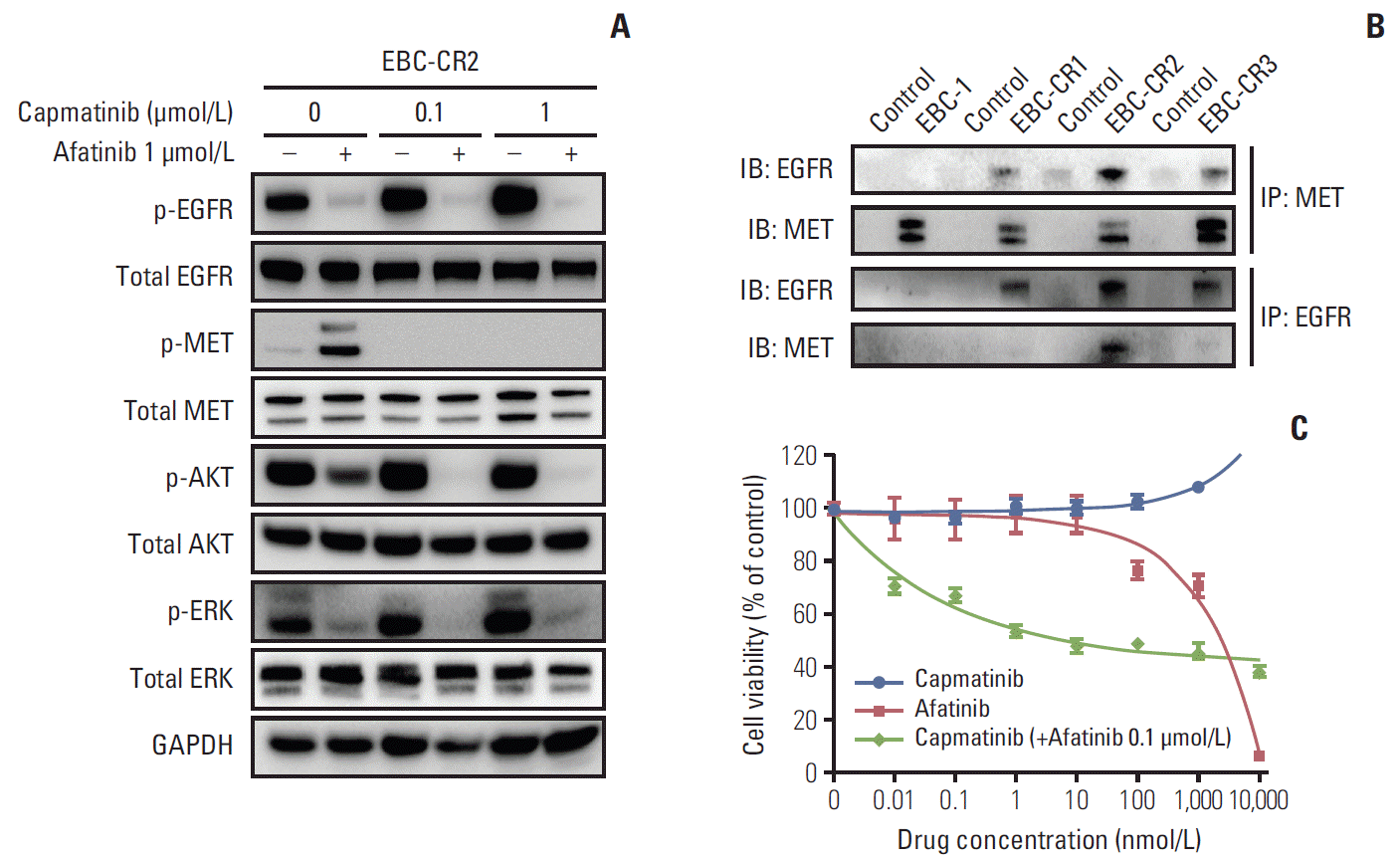
3. Increased MET and EGFR heterodimerization can cause acquired resistance to capmatinib
In contrast to EBC-CR1 cells, in EBC-CR2 cells, the combined blockade of MET and EGFR kinase significantly abolished phosphorylation of the downstream signaling molecules, AKT and ERK1/2 (Fig. 3A). While activation of both MET and EGFR kinase was diminished in the parental cell line, EGFR phosphorylation was increased in EBC-CR2 cells by capmatinib treatment and MET phosphorylation was increased by afatinib. Intriguingly, EBC-CR2 cells displayed more dominant MET-EGFR heterodimers than the parental cell line (Fig. 3B). To determine the efficacy of combined treatment with capmatinib and afatinib, we performed the cell viability assay to compare the effect of capmatinib, afatinib, and their combination in EBC-CR2 cells. We observed that the IC50 was lower in the cells treated with the combination compared to either compound alone, which suggested that capmatinib and afatinib synergistically inhibited cell proliferation (Fig. 3C). These results indicated that dual blockade of MET and EGFR obstructed the MET-mediated EGFR survival signal caused by MET and EGFR heterodimerization. Accordingly, the MET-EGFR heterodimer is a much more dominant form of activated EGFR kinase, which is the main survival signal in EBC-CR2 cells.
4. Acquired resistance to EGFR TKI beyond capmatinib resistance
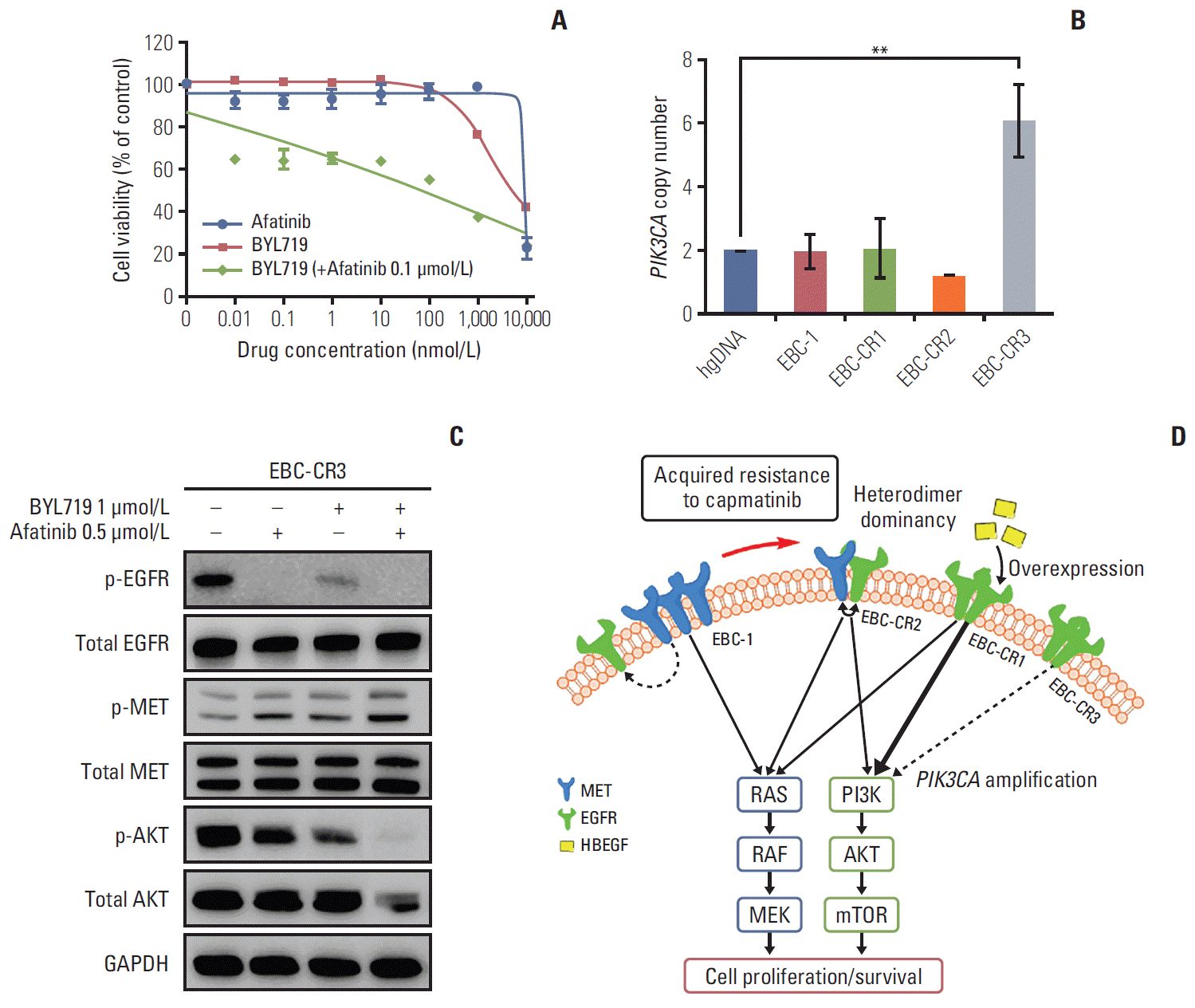
Despite continuous exposure of the EBC-CR3 cell line (derived from EBC-CR1 cells) to capmatinib but not EGFR-TKI resulted in the loss of sensitivity to afatinib in the cell viability assay (Fig. 4A). In Western blot, EBC-CR3 showed a complete block of EGFR or MET activation, but persistent AKT phosphorylation after afatinib treatment (S5B Fig.). We performed qRT PCR and Sanger sequencing to investigate the mechanism underlying EGFR-dependent resistance to afatinib. We found that PIK3CA mRNA expression was increased and gene copy number was amplified in EBC-CR3 cells, which was unlike what we observed in the other cell lines (Fig. 4B, S5C Fig.); however, we did not find any hot spot mutations in PIK3CA (data not shown). PIK3CA amplification was observed frequently and might play a pathogenic role in lung cancer, especially squamous cell carcinoma [17]; thus we hypothesized that PIK3CA amplification is acquired resistant mechanism to capmatinib. To confirm this hypothesis, we tested the efficacy of BYL719 on PIK3CA inhibition in EBC-CR3 cells, because this molecular target of BYL719 had been reported previously. [18] As a result, we showed resistance after a single treatment with BYL719; however, the cells with concurrent EGFR overexpression were sensitive to BYL719 in the presence of afatinib in the cell viability assay (IC50 for BYL719, 140.84±19.65 nmol/L) (Fig. 4A). Although EBC-1 and EBC-CR2 cells were resistant to combined treatment of BYL719 and afatinib, it showed additive effect on EBC-CR1 cells (S6A Fig.). In addition, the combination of BYL719 with afatinib effectively inhibited phosphorylation of AKT, despite the persistence of the MET signal (Fig. 4C), and inactivation of RAS/MAPK pathway (data not shown). The combined treatment incompletely inhibited AKT phosphorylation on EBC-1 and -CR2 (S6B Fig.). Although one or more colony might be formed in EBCCR3 cells, there was no other genetic modification in other colonies except PIK3CA amplification. These results demonstrated that wild-type PIK3CA amplification could be an additional mechanism of resistance to EGFR-TKI in capmatinib-resistant cancers; however, the combined therapy effectively overcame the resistance to TKIs in a MET-independent manner in the presence of additional genetic alterations.
Discussion
In this study, we observed EGFR mRNA overexpression without the presence of an EGFR activating mutation or amplification, as described above. These findings suggested that common resistance to capmatinib occurred via activation of signaling pathways outside of MET kinase, rather than via genetic alteration of specific genes. Furthermore, partial or complete engagement of the EGFR signal was one resistance mechanism against capmatinib, which indicated a preferable alternative signaling pathway in MET-dependent cancer cells.
Previous clinical research reported de novo MET amplification in NSCLC patients. MET amplification may be druggable targets for MET-TKIs on the basis of preclinical and clinical findings [19-21]. Therefore, preclinical and clinical trials of MET-TKIs have been reported for several cancers. Capmatinib, which is currently in clinical trials, is a very selective and potent MET-specific agent for MET-dependent cancer [6,13,15]. However, rapid development of acquired resistance to MET-TKIs has been crucial limitation to efficacy. Nonetheless, only a few resistance mechanisms of MET-TKIs have been elucidated up until now. Therefore, it is important to identify the underlying mechanisms of MET-TKIs resistance through molecular studies and establish effective therapeutic strategies to manage acquired resistance. Although an in vivo model is important, in vitro cancer cell lines can provide a valuable model system to investigate resistance mechanisms. Here, we have used this in vitro strategy to identify molecular resistance mechanisms of capmatinib in MET-amplified NSCLC cell lines.
MET positively regulates EGFR signal pathway via multiple mechanisms, so EGFR phosphorylation decrease by selective MET-TKI, capmatinib in MET-amplified NSCLC cell lines [6]. The EGFR signal has been reported as a preferable bypass mechanism for MET kinase, and the combination of EGFR-TKI with MET-TKI renders resistant cells sensitive, as described in several studies [22-24]. In addition, the clinical evidence of EGFR co-amplification as a resistance mechanism to other MET kinase inhibitor existed in MET-amplified esophagogastric cancer [25]. One of our resistant cell lines (EBC-CR2) also was sensitive to the combined treatment. Interestingly, phosphorylation of EGFR in the EBC-CR2 cell line was increased by capmatinib in a dose-dependent manner, in contrast to the parental EBC-1 cell line. Therefore, we assumed that MET and EGFR kinase may be activated via heterodimer formation and the ability of the main cell signaling pathway to freely alternate between MET and EGFR. The EGFR pathway was mainly activated in the presence of MET-TKI and the MET pathway was activated in the presence of EGFR-TKI because of heterodimer interactions. EBCCR2 cells displayed the most MET-EGFR heterodimerization compared to the parental and other resistant cell lines, as previously mentioned. EGFR heterodimerization as resistant mechanism has been reported in some RTK inhibitors [26,27], but not in MET-TKI. These results suggest that heterodimer can be acquired mechanism of MET-TKI for the first time. Presumably, physical interaction between MET and EGFR might facilitate the formation of MET-EGFR heterodimer or trans-phosphorylation [28]. In addition, effective suppression of cell proliferation in response to combined treatment supports combined therapy with MET and EGFR inhibitors as a potential therapeutic strategy for MET-TKI acquired resistance in cancer patients.
EGFR phosphorylation was inhibited by capmatinib in the parental EBC-1 cell line, but not in the EBC-CR1 cell line (Fig. 2C). This result suggested that the EGFR signal of EBC-CR1 was independent of the MET signal. The survival signal in EBC-CR1 cells had completely shifted from MET to EGFR, because a single treatment with afatinib effectively inhibited downstream signaling and cell proliferation. Intriguingly, EBC-CR1 cells, which were completely EGFR-dependent, became resistant to afatinib via PIK3CA amplification by culture in continuous high concentrations of capmatinib as EBCCR3 cells (Fig. 3A and B). Genetic alteration of PIK3CA frequently co-occurs with other oncogenic alterations including MET [29]. For example, concurrent PIK3CA mutation and the MET exon 14-skipping mutation, which results in constitutive activation of MET, result in primary resistance to MET-TKIs [30,31]. In the case of EBC-CR3 cells, PIK3CA amplification might be an important mechanism of acquired resistance to EGFR-TKI beyond MET-TKI resistance, which was demonstrated by the independence of EBC-CR1 cells from the MET kinase signal.
Despite exposure to similar final concentrations of capmatinib (EBC-CR2, 2.2 μmol/L; and EBC-CR3, 2.4 μmol/L), EGFR/MET co-dependent EBC-CR2 cells consistently showed sensitivity to the combination of capmatinib with afatinib. In contrast, EBC-CR3 cells acquired additional resistant mechanism of PIK3CA amplification. Based on this observation, a possible explanation for resistance is that EGFR-MET heterodimerization increased the cells’ genetic stability or had a higher threshold concentration to induce a genetic change; thus, a genetic alteration was not required in EBC-CR2 cells. Anticancer drugs promote chromosomal instability by inducing fragile sites in chromosomes that lead to amplification events [32]. In addition, we observed that EBC-CR3 cells had less DNA repair-related gene expression than EBC-CR1 (Table 2, S3A Fig.). AKT overactivation might suppress DNA repair in EBC-CR3 cells like a previous study [33]. However, it is unknown whether DNA repair-related genes clearly contribute to capmatinib resistance and further study may be necessary to investigate the role of DNA repair genes as a capmatinib resistance mecahnism. Capmatinib had less of an effect on the unconstrained survival signal change in response to RTK inhibitors in EBC-CR2 cells. In contrast to EBC-CR2 cells, EBC-CR1 cells relied on ligand-dependent activation, which was less stable resistant mechansism to kinase inhibitor than heterodimerization. Therefore, a secondary event was required to increase the stability of the cells against potent MET inhibition, and shift the cells toward a MET-independent survival signal. Afatinib plus BYL719 effectively inhibit PIK3CA-amplified EBC-CR3 cells by the down-regulation of phosphorylated AKT signal that is a downstream effector of PI3K signal. Taken together, although treatment schedules or drug concentrations were similar, in vitro drug-resistant models showed heterogeneous resistance mechanisms in various resistant clones [34] like patients who have heterogenous resistance mechanisms [35].
In conclusion, our findings suggested that a single cell line can develop several types of EGFR-dependent mechanisms of resistance to MET-TKIs in MET-amplified NSCLC cell lines. One mechanism involved an increase in ligand and receptor expression. Indeed, we found increased EGFR and HBEGF mRNA expression or MET-EGFR heterodimer formation, which resulted in sensitivity of EBC-CR2 cells to combined therapy, as mechanisms to promote acquired resistance. The most interesting finding was that capmatinib treatment resulted in a shift from capmatinib resistance to afatinib resistance via PIK3CA amplification.
Because cancers become resistant to MET-TKI in clinical settings, it is important to understand TKI-resistance mechanisms in preclinical and clinical studies. Therefore, our results highlighted the relationship between MET, EGFR and additional altered oncogenes such as PIK3CA in the context of therapeutic strategies involving kinase inhibitors in capmatinib-resistant NSCLC cells.
Future efforts should be directed to prove the resistant mechanisms through an in vivo model as well as clinical data from MET-amplified NSCLC that acquires resistance to capmatinib (NCT02414139).
 XML Download
XML Download