

 PDF
PDF Citation
Citation Print
Print


Introduction
Head and neck squamous cell carcinoma (HNSCC) is the sixth most common malignancy worldwide, and is usually curable, if diagnosed early. Unfortunately, patients often present with advanced disease that is incurable or requires aggressive treatment, which results in functional disability, dismal prognosis and high mortality. Low survival outcomes in combination with significant toxicity of current treatment strategies emphasize the necessity for novel therapeutic modalities. Until recently, the only targeted therapy in HNSCC was cetuximab, a monoclonal antibody against epidermal growth factor receptor, which has shown a response rate of 10% to 15% in the patients with recurrent or metastatic disease [1]. However, there is no validated biomarker for predicting cetuximab efficacy, which dampens the precise selection of patients. Anti‒programmed death 1 agents including pembrolizumab and nivolumab were recently approved for HNSCC that is refractory to platinum-based therapy. However, the presence of programmed death-ligand 1 (PD-L1) on tumor cells did not satisfactorily predict response, with 22% of PD-L1 positive patients responding vs. 4% of PD-L1 negative patients responding [2]. Therefore, more effective treatment strategies for personalized treatment of HNSCC are urgently needed.
Next-generation sequencing (NGS) of tumors has greatly expanded our understanding of genetic profiles, and several studies have found novel genetic alterations in HNSCC [3-6]. However, these studies were performed retrospectively in surgical specimens without incorporated clinical data on the response to therapy. Although potentially targetable genetic alterations such as PIK3CA, epidermal growth factor receptor (EGFR), and fibroblast growth factor receptor (FGFR) mutations have been identified, functional studies to validate the roles of such mutations as biomarkers remain scarce.
Herein, we describe our implementation of a precision medicine approach in 93 patients with HNSCC. This is a feasibility study of “Translational biomarker-driven umbrella project for head and neck and esophageal squamous cell carcinoma (TRIUMPH)” study by the Korean Cancer Study Group (NCT03292250) (S1 Fig.). TRIUMPH is the first, prospective, biomarker-driven umbrella trial for patients with HNSCC, consisting of multiple targeted therapies including phosphoinositide 3-kinase (PI3K) inhibitor, pan-HER inhibitor, FGFR inhibitor and CDK4/6 inhibitor. Patients without actionable targets are to be allocated into an immunotherapy arm. Before the start of TRIUMPH study, we conducted this feasibility study in which tumors and matched blood samples were analyzed by multiplexed targeted NGS assays. The objective of this study is to examine the feasibility of implementing NGS to guide treatment in HNSCC patients, and to find the associations between somatic alterations and clinical outcome.
Materials and Methods
1. Patients and data collection
Pretreatment tumor tissues (somatic) and matched normal DNA (germline) from prospectively recruited patients with HNSCC were obtained between 2016 and 2017 under the approval of Institutional Review Board of 19 institutions in Korea. HNSCC patients with initial stages 1-4 were included in this study. Clinicopathological data were collected from patient charts in accordance with an IRB-approved protocol. Clinical information including age, sex, anatomic site of tumor, tobacco and alcohol use, clinical stage, treatment history, and survival data were collected.
2. Targeted sequencing of tumors
Genomic DNA was isolated from formalin-fixed paraffinembedded (FFPE) samples using the QIAamp DNA FFPE Tissue Kit (Qiagen, Hilden, Germany) for the targeted sequencing of 244 head and neck cancer-related genes selected based on a literature search (S2 Table). The genomic regions of the 244 genes were captured by the customized SureSelectXT Target Enrichment library generation kit (Agilent, Santa Clara, CA), and sequenced using the Illumina HiSeq 2500 platform with a depth of coverage > 1,000×.
3. Variant calling and functional annotation
By default, base quality trimming for short reads from the targeted sequencing was performed using Sickle [7]. Filtered reads were mapped to the human reference genome (GRCh-37/hg19) using Burrows-Wheeler Aligner [8]. All reads that were mapped with < 23 mapping quality were discarded. The aligned reads (BAM file) were further processed with the Genome Analysis ToolKit v3.5, including MarkDuplicate, Local Realignment, and Base Quality Score Recalibration [9]. Initial somatic mutations candidates were called by MuTect ver. 1.17 with a default parameter [10]. Somatic insertions/deletions (indels) were called by Varscan2 ver. 2.3.7 with somatic p < 0.05 [11]. From the initial call set, FoxoG artefacts were removed using the in-house Python program ver. 3.6 to discard skewed read-orientation variants [12]. The functionality of final high confidence variants was annotated with ANNOVAR software [13], including the consequences, predicted impacts and reported allele frequencies in population. In particular, non-rare variants (minor allele frequency > 0.05) were discarded to retain only pathogenic variants. Finally, the clinical interpretation of targeted therapy was annotated using the CIVIC [14] and DoCM [15] databases. Copy number alterations (CNAs) were called using CNVkit [16] for targeted deep sequencing. To reduce ambiguity from individual variations, all normal samples were pooled and used as a control. Of the initial CNA calls, genes with ≥ 4 and 0 measured copy numbers were considered amplified and deleted, respectively, to secure high confidence. To visualize the overall landscape of mutations, ‘Oncoprint’ was drawn using the R package ‘ComplexHeatmap’ of R ver. 3.4. Lollipop plots were drawn for frequently mutated genes using MAFtools to check the recurrence of genomic loci with variants.
4. Nanostring assay
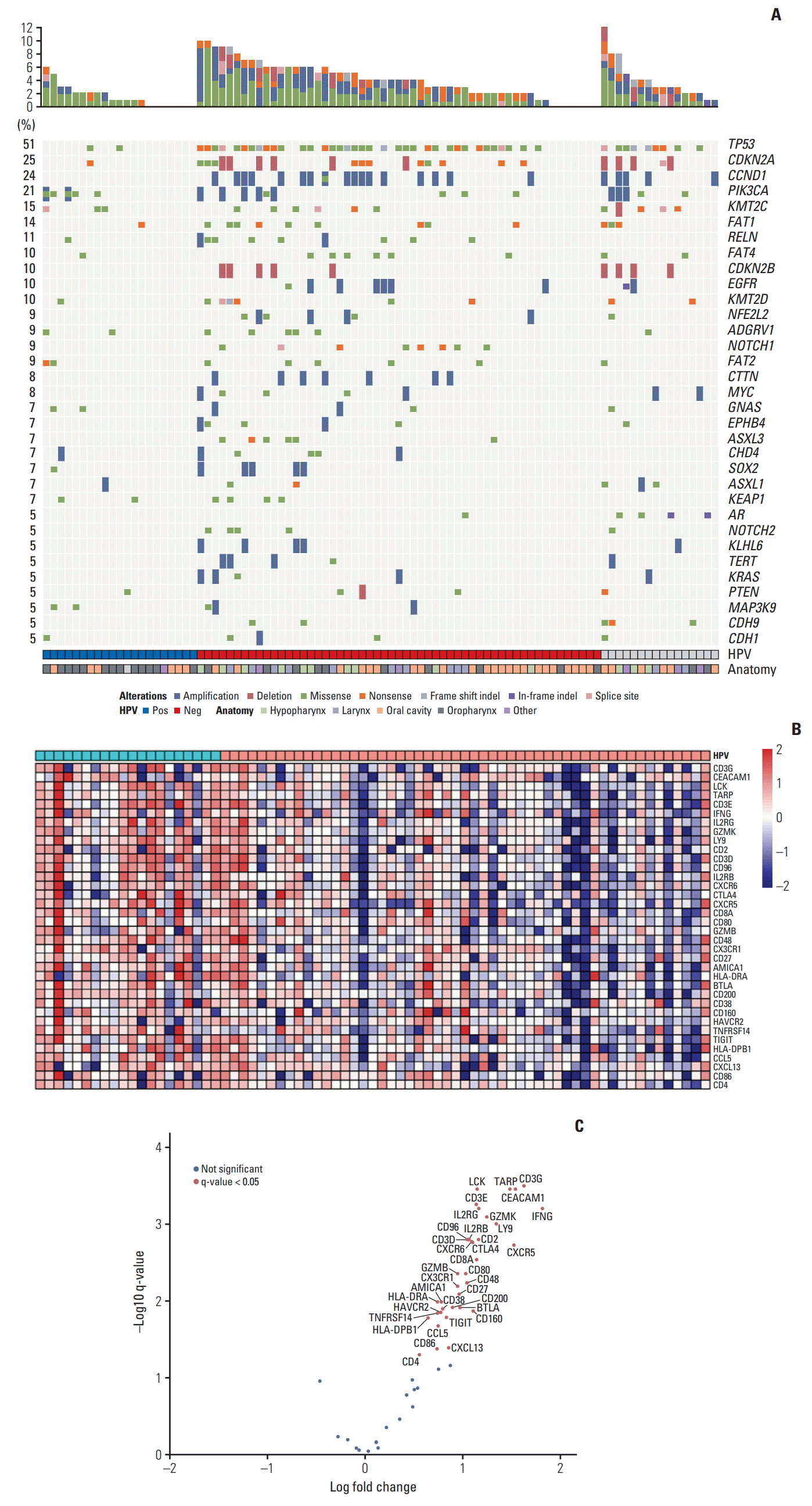
Total tumor RNA was isolated using the RNeasy kit (Qiagen). The nCounter Analysis System (Nanostring Technologies, Seattle, WA) was used to screen for the expression of 55 immune-related genes. Counts were normalized to internal controls and reference genes using the nSolver software ver. 3.0. We obtained gene expression data for 94 tumour samples, among them 8 with average expression levels of less than 10 were filtered out. The NanoStringNorm package of R was used for normalization [17]. We selected housekeeping. geo.mean for normalizing the samples or RNA contents. Differentially expressed genes between human papillomavirus (HPV)–positive and HPV-negative samples were identified by the glm.LRT function in the NanoStringDiff [18] package of Bioconductor. A volcano plot was drawn by using the ggplot2 package of R. The complete list of 55 immune-related genes is shown in S3 Table.
5. Statistical methods
All statistical analysis was performed using the R, Python Scipy package and SPSS ver. 23.0 (IBM Corp., Armonk, NY) software. To test group-specific enrichment of genomic variants, Fisher exact test was applied to each called variant, followed by the p-value cutoff of 0.05. Tumor mutation burden (TMB) was measured by the number of missense mutations per megabase (Mb) within the range of the targeted capture region. The numbers of mutations per Mb between HPV-positive and HPV-negative groups were compared using Student’s t-test. Progression-free survival (PFS) and overall survival (OS) were estimated using the Kaplan-Meier method; differences between groups were compared using the logrank test. In groups of unbalanced sizes, the standard asymptotic log-rank test is often replaced by its corresponding permutation test; alternatively, the distribution under the null hypothesis is approximated via Monte Carlo resampling. Here, we used empirical p-values from 10,000 replicates by using the log-rank test function in the coin package of R. Two-sided p-values of < 0.05 were considered significant.
Results
1. Clinical characteristics
A total of 93 patient tumors were included in 75 men and 18 women. Clinical data are summarized in Table 1; the median age of all patients was 59 years (range, 28 to 80 years) and 39 patients (42%) had stage 4 disease at initial diagnosis. Sixty-seven patients (72%) had smoking history and 59 patients (63%) had alcohol history. HPV status was known in 76 patients (82%), of whom 20 (22%) were positive. Sixty-eight patients (73%) had received prior surgery, and among patients who received surgery, 47 patients experienced recurrence: 14 (29%) with locoregional recurrence and 33 (71%) with distant metastasis. Surgery or radiotherapy was performed for locoregional recurrence, and systemic chemotherapy was performed for metastatic disease. For the whole cohort, the median PFS and OS were 12.5 months (95% confidence interval [CI], 10.2 to 14.8) and 70 months (95% CI, 57.4 to 84.4), respectively, with a median follow-up of 20 months. Patients with HPV-positive oropharynx cancers (n=16) showed a better 2-year OS rate than HPV-negative patients (n=10) (31% vs. 10%, respectively), although the difference was not significant owing to the small number of cases.
2. Overview of somatic mutations in HNSCC
A total of 2,315 somatic single nucleotide variations (SNVs) and 19 indels were identified from the targeted sequencing of the 92 tumors, which corresponds to a rate of 3.64 SNVs per 1 Mb. We found that TP53 was the most frequently mutated gene (n=47, 51%), followed by CDKN2A (n=23, 25%), CCND1 (n=22, 24%), and PIK3CA (n=19, 21%) (Fig. 1A). As expected, smokers displayed a significantly higher TMB than non-smokers (4.16/Mb vs. 3.12/Mb, p=0.04) (S4 Fig.).
3. Comparison of HPV-positive vs. HPV-negative tumours
Of 92 patient tumors, 76 tumors (82%) had known HPV status and we compared molecular landscape of HPV-positive and HPV-negative tumors. TMB counts were higher in HPV-negative than HPV-positive tumors, although the difference was not significant (4.16/Mb vs. 3.12/Mb, p=0.150) (S5 Fig.). TP53, CDKN2A, and CCND1 gene alterations were significantly more frequent in HPV-negative tumors (Fig. 1A). As described previously, we observed TP53 mutations among HPV-negative tumors at higher rates than HPV-positive tumors (65.5% vs. 9.5%, p < 0.001). Inactivating mutations such as CDKN2A and CDKN2B deletions (n=6), and CCND1 amplification (n=17) were exclusively identified in HPV-negative tumors. We also noted HPV-negative specific genetic alterations in receptor tyrosine kinases (RTKs) including EGFR, FGFR1/3, and platelet-derived growth factor receptor A (PDGFRA), which was consistent with a previous study [5]. PIK3CA mutations were more commonly found in HPV-positive tumors (23.8% vs. 16.4%, p > 0.05). Comparison of immune signatures between HPV-positive and HPV-negative tumors via nanostring assay revealed that HPV-positive tumors were significantly enriched with immune-related genes. HPV-tumors harbored higher levels of immune activation: specifically, CD3 (p=6.0×10–6), CECAM1 (p=4.9×10–5) and IL2R (p=6.9×10–5) expression (Fig. 1B and C).
4. Clinical correlation
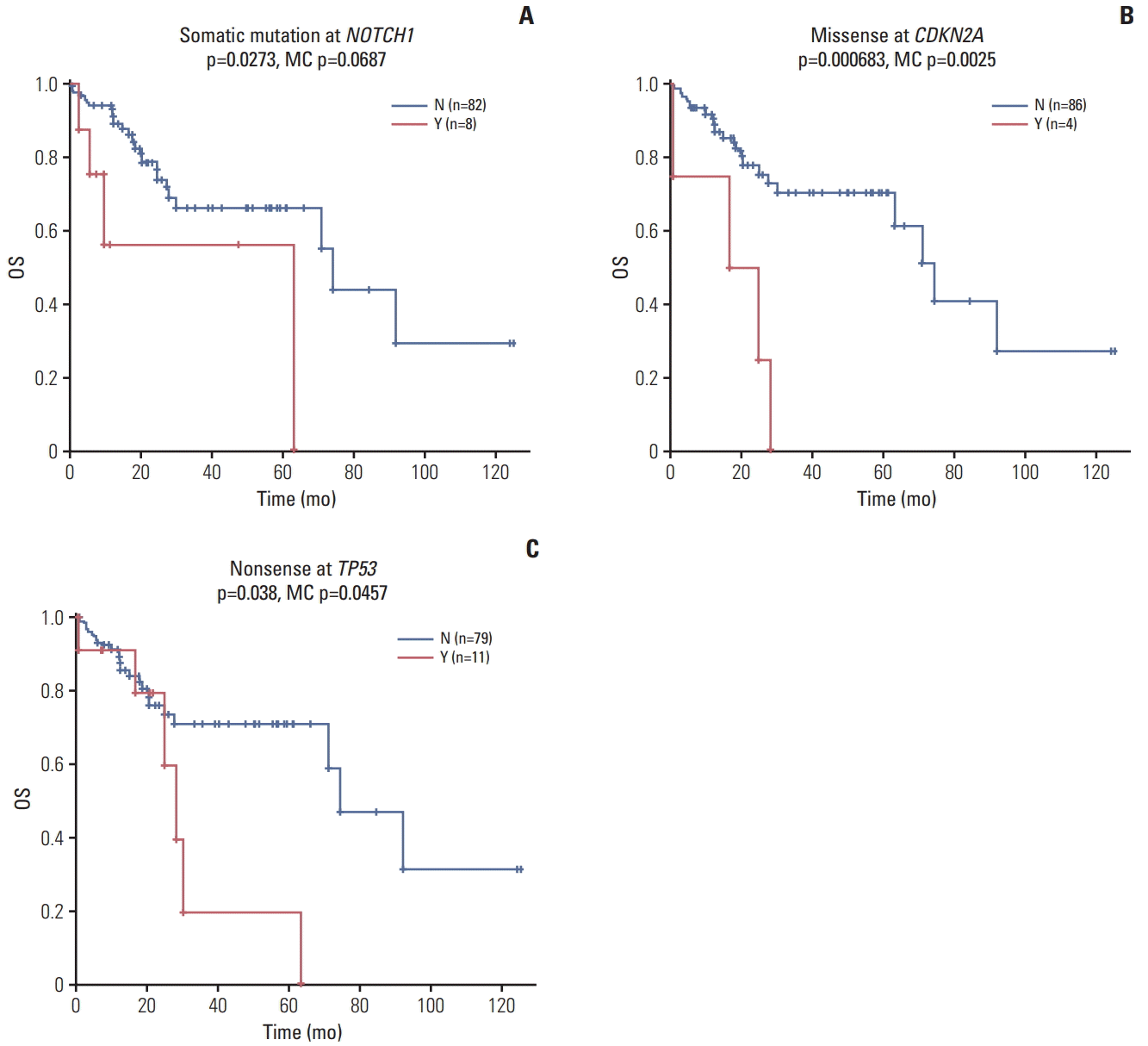
We performed an exploratory analysis to correlate gene alterations (SNVs and CNAs) with survival (Fig. 2). In 90 patients with available survival data, genomic events associated with poorer OS were mutations in NOTCH1 (p=0.027), CDKN2A (p < 0.001), and TP53 (p=0.038). The association between CDKN2A, TP53 mutations and poor OS was consistent with a previous analysis of The Cancer Genome Atlas (TCGA) database. CNAs were not associated with any gene alterations. In contrast to a previous report [19], PIK3CA amplification was not associated with worse OS (S6 Fig.).
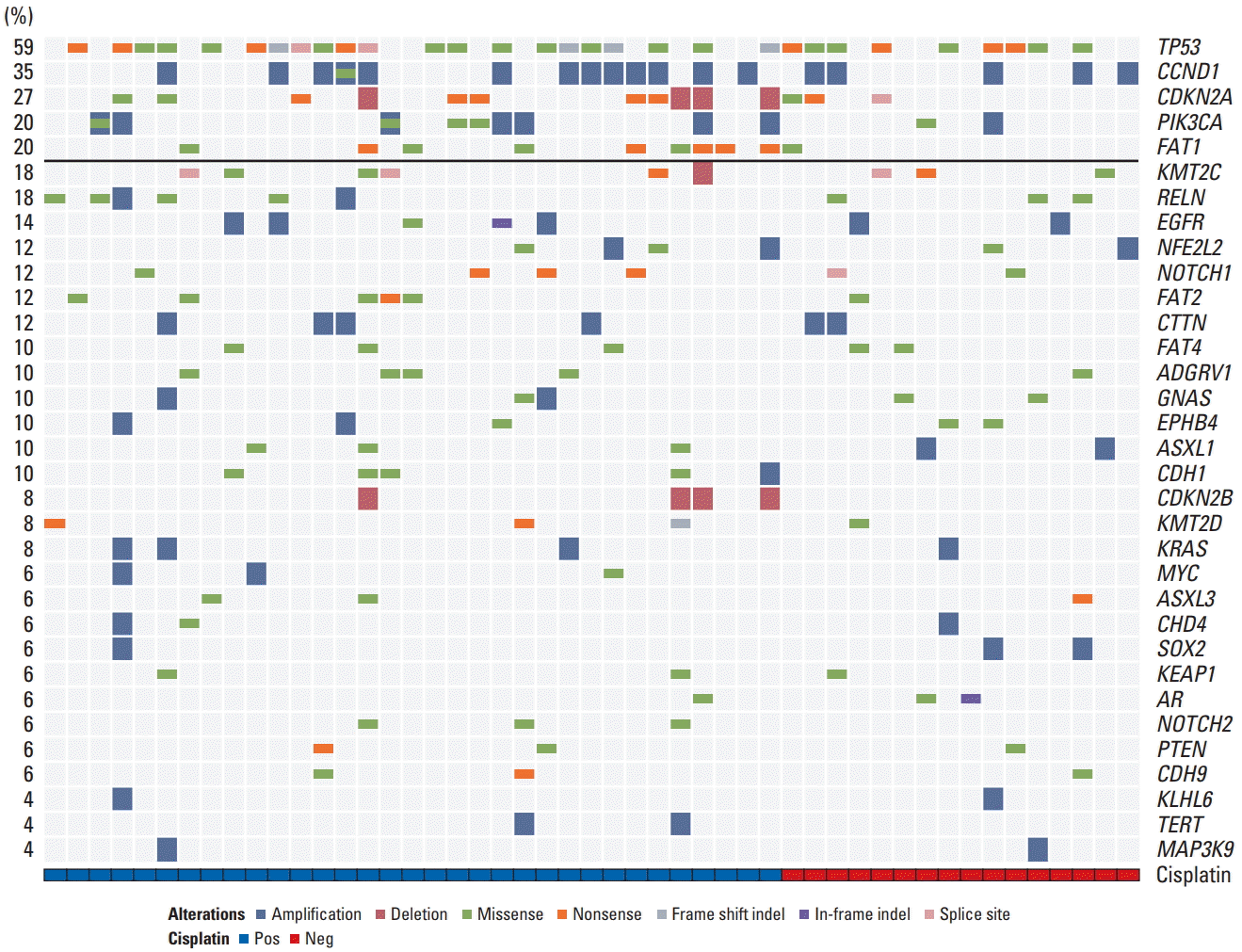
Next, we analyzed gene alterations associated with cisplatin resistance by classifying patients who received cisplatin-based chemotherapy into responders and non-responders. According to Response Evaluation Criteria in Solid Tumors (ver. 1.1), responders were patients who showed complete response, partial response or stable disease to cisplatin-based chemotherapy, whereas non-responders were those with progressive disease [20]. Among 54 evaluable patients, 38 (70%) were responders, and 16 (30%) were non-responders. FAT1 gene mutations (5 missense and 5 nonsense) were highly enriched in cisplatin responders compared to non-responders (p < 0.05) (Fig. 3, S7 Fig.).
5. Targetable mutations and copy-number aberrations
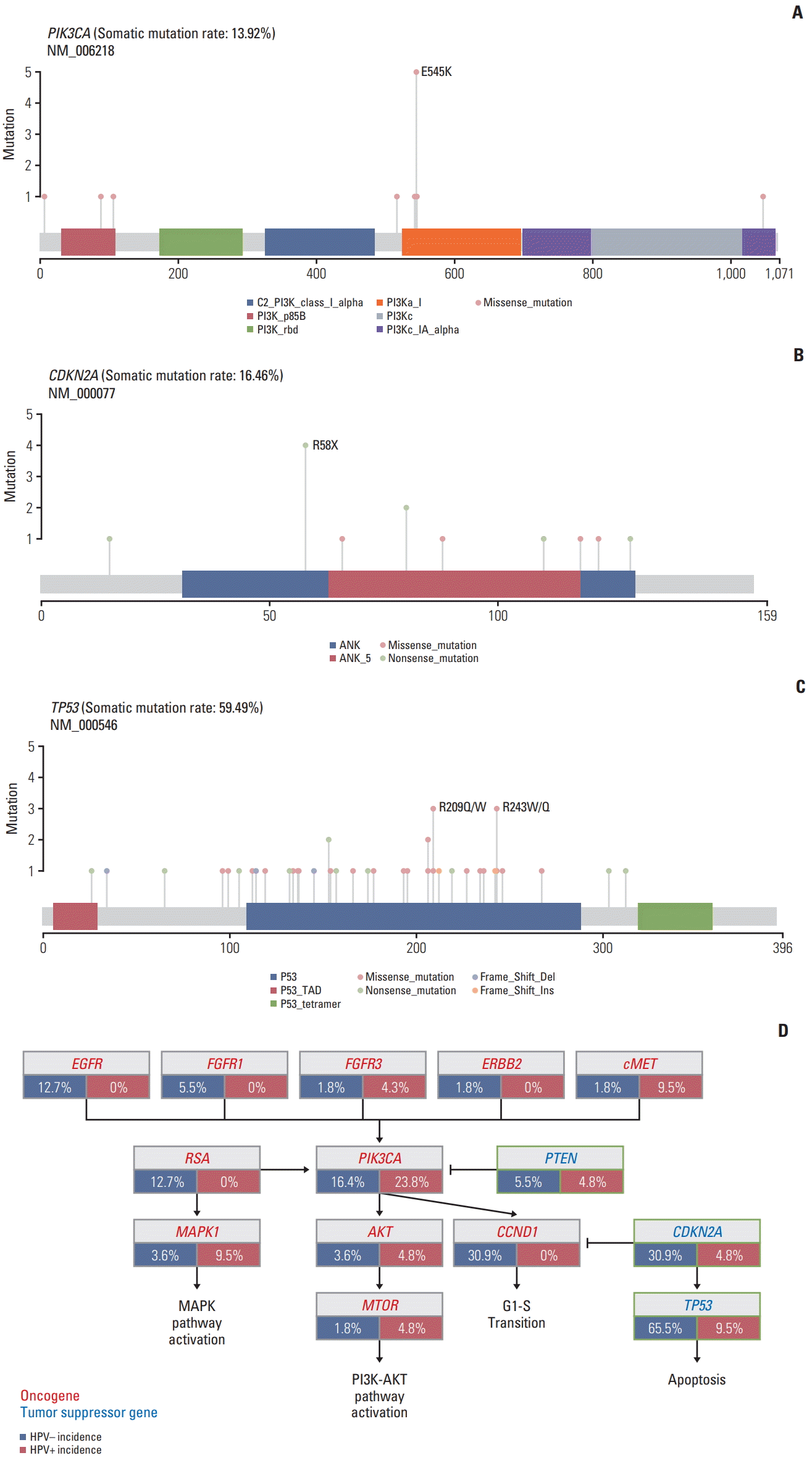
We identified potentially targetable mutations in PIK3CA and CDKN2A. An established canonical mutation, PIK3CA E545K missense mutation were identified in five patients (5%) (Fig. 4A), while CDKN2A R58X nonsense mutation was identified in four patients (4%) (Fig. 4B). TP53 inactivating mutations (R209Q/W, R243W/Q) that cause cell cycle deregulation occurred in eight patients (5%) (Fig. 4C).
Fig. 4D summarizes the deregulated signaling pathways. Among RTKs, EGFR and cMET alterations were frequent, followed by FGFR3, FGFR1, and ERBB2. Among downstream targets of the RTKs/RAS/PI3K pathway, PIK3CA dominated with occasional MAPK1 and MTOR mutations. RAS and MAPK1 alterations occurred in 12.7% and 13.1% of patients, respectively. Alterations in tumor suppressors, TP53 and CDKN2A were notable in HPV-negative tumors, which were consistent with a recent report [21]. Overall, alterations in genes involved in cell death and PIK3CA/AKT/MTOR pathway were predominant.
Discussion
Our umbrella trial suggests that using NGS for determining treatment strategies for patients with HNSCC is feasible, and that translating genomic data into clinical care is attainable. The most common genomic alterations (TP53, PIK3CA, CCND1, and CDKN2A) were identified at frequencies consistent with investigations of TCGA. Previous studies have characterized mostly surgically resected HNSCC samples, with a limited portion of HPV-positive samples. TCGA study, which is the largest cohort to date (n=279) is comprised of surgically resected oral cavity or laryngeal squamous cell carcinoma patients, and treatment and survival data were limited [5]. Recently, Seiwert et al. [22] reported a large number of HPV-positive tumors, where they included 51 (42.5%) HPV-positive patients in a total of 120 patients. Consistent with our finding, the mutational burdens in HPV-positive and -negative tumors were similar, while FGFR2 aberrations were exclusively identified in HPV-negative tumors.
Our study emphasizes how the application of NGS may be used as a prospective, master protocol tailored to each patient’s genotype. The turnaround time from patient sample collection to NGS results was within 4 weeks, which is timely for patient enrolment. Similarly, another study recently found it feasible to incorporate NGS into the clinical care of HNSCC patients [19]. Patients who received targeted therapy matched to their genotypes achieved a higher objective response rate than patients unmatched to therapy. However, they used two different NGS platforms with inconsistent mutation rates and actionable alterations. Additionally, the MOSCATO-01 trial showed that genomic analyses of 199 patients with advanced cancers produced improved outcomes with matched targeted therapy [23]. The ongoing NCI-MATCH trial is currently assessing whether molecular markers can predict response to targeted therapies in patients with advanced cancer [24] and the results are awaited.
PI3K pathway aberrations are potential therapeutic targets in HNSCC patients. Prior studies identified that PIK3CA mutation or amplification was associated with various clinical outcomes. One study reported that PIK3CA amplification was associated with significantly decreased PFS, whereas PIK3CAmutation was not [19]. Another study demonstrated that PIK3CA mutations were correlated with poor prognosis in HPV-negative, locally advanced HNSCC [22]. A preclinical study also reported that patient-derived PIK3CA mutant HNSCC tumor grafts are potentially sensitive to PI3K/MTOR inhibitors [25]. In our cohort, patients with a PIK3CA hotspot mutation (E545K) will be treated with the PI3K pathway inhibitor (BYL719).
Deletion of CDKN2A or amplification of CCND1, which induces sustained CDK 4/6 activation, occurred at 27% and 22%, respectively, which were comparable to such cell-cycle related gene aberrations found in other studies [5,22]. Preclinical or clinical data regarding the activity of CDK inhibitor in HNSCC is limited, but our prospective trial may solve which genotypes will benefit from treatment with CDK inhibitors.
FAT atypical cadherin 1 (FAT1) was significantly enriched in cisplatin responders. FAT1 gene has been reported to be associated with various types of cancer, including HNSCC [5,26]. FAT1 gene acts as a tumor suppressor, in which loss-of-function activates Wnt pathway and tumorigenesis [27]. Recently, FAT1 mutation was significantly associated with better OS in HPV-negative patients from both the TCGA cohort and the International Cancer Genome Consortium (ICGC) data cohort [28]. The functional impact of the FAT1 mutation identified in our study requires further investigation to determine its role as a prognostic or predictive biomarker.
In our study, immune signatures were highly enriched in HPV-positive tumors, consistent with a previous finding that HPV-positive tumors have a distinct immune phenotype, characterized by more immune cell infiltration and higher levels of CD8+ T-cell activation [29]. As ongoing checkpoint inhibitor trials (NCT02105636, NCT01848834) showed promising preliminary activities in HNSCC patients, improved outcome in HPV-positive patients may be related to their immunophenotype [30,31].
The accuracy and fidelity of genomic analysis are critical; therefore, false-positive or false-negative genomic variants should be carefully avoided. To that end, several technical issues were noted in our study. First, the often inevitable low tumour cellularity in samples, owing to normal cell contamination, has a negative effect on the accuracy of calling of SNVs and CNVs [32]. We found that, among 92 samples, 14 (15%) and five (5%) obtained via core needle biopsy and excision, respectively, showed low tumor cellularity (~30%). As CNV analysis is directly affected by reduced cellularity, CNVs with ambiguous analysis scores may require confirmation using alternative methods. Second, sequencing artefacts can appear in every step of the NGS pipeline, which complicates the differentiation between true vs. false variants. We observed an abnormally excessive number of lowlevel somatic mutations in a few samples (mutation-rate/Mb > 100), which could only be removed using an oxoG filtering program [12]. Such false variants can distort the overall distribution of somatic mutations and their relative burdens, and should be specially inspected via advanced bioinformatics analyses. Third, whole-exome or targeted sequencing for identifying CNVs remain secondary options, as more sensitive methods such as whole-genome sequencing or specialized array-based methods are widely unavailable. As targeted sequencing based CNV analysis generally performs better in a larger cohort, size and sustainability of clinical trials should be considered when they are designed. Moreover, active participation of genome analysis experts is highly recommended to manage such technical issues.
In conclusion, our large-scale targeted sequencing of HNSCC patient samples identified potentially targetable alterations. Further prospective validation of NGS based molecularly targeted treatment is highly warranted.
 XML Download
XML Download