

 PDF
PDF Citation
Citation Print
Print


Introduction
Colorectal carcinoma is the second most common cause of cancer-associated death worldwide [1,2]. Though half of colorectal cancer patients showed a positive initial response that benefited from the successful surgery or multimodal treatment [3], many patients had worse treatment efficiency and dismal prognosis which were considered to be induced by visceral metastasis [3]. Moreover, the 5-year survival for patients with metastasis was lower than those who had no metastasis [4]. However, the specific molecular mechanisms underlying the development of metastasis in colorectal cancer remain unclear [5]. Hence, identifying the drivers that influence the distant metastasis may lay an important foundation to make progress in the diagnosis and therapy for colorectal cancer patients.
Given the potential role on clinical treatment, a better understanding of the mechanism of distant metastases in colorectal cancer is critical. Increasing evidences reveals cancer metastasis depends on various curial factors, including up-regulation of integrins [6] and E-cadherin [7,8], epithelial–mesenchymal transition (EMT) progression of tumor cells [9], hypoxia [10], tumor microenvironment [11] and so on [12]. EMT was initially reported as a tightly regulated lineage change during gastrulation, neural crest delamination, heart valve formation, and embryonic development. Recently, increasing attention has been attracted for the important role of EMT in cancer metastasis [13]. A lot of molecules are responsible for the EMT in the tumor cells, including inflammatory cytokines, growth factors and numerous transcription factors. In recent years, the tumor microenvironment has received attention as an important determination of cancer cells metastasis, among which tumor-associated macrophages (TAMs) have been reported to play a prominent functional role in cancer behaviors [14]. TAMs are the major inflammatory component in many tumor microenvironments, which affect different aspects of tumor behaviors [15], including participation in tumor growth in several ways, such as transforming growth factor-β (TGF-β) secretion [16], remodeling of tumor microenvironment, inflammation suppression [17] and so on. TGF-β is one of the major endogenous regulators of cell growth, which shows enhanced expression in various human cancers, including breast cancer, colorectal cancer, pancreatic cancer and so on [18]. TGF-β participates in multifarious tumor progress, including potent immunosuppressive effects, EMT progression of tumor cells [19]. Increasing evidences demonstrated that TAMs facilitate distant metastasis in various tumors [20], including lung cancers, breast cancer and so on. However, the specific mechanism of metastasis induced by TAMs is still unclear and it is a challenge to inhibit the metastasis of colorectal cancer in clinic therapy.
In our study, our aim is to reveal the expressional profile of TAMs in the colorectal cancers and investigate its potential role in the metastatic progress. Moreover, finding the underlying mechanism involved in the colorectal cancer metastasis may lay an important foundation for the clinical treatment. We examined enriched TAMs in tumor tissues from colorectal patients with high metastasis, which was proved to facilitate tumor cells migration and neoplasm metastasis. Furthermore, our study reveals that TAMs promote the progression of EMT in colorectal cancer cells through the secretion of TGF-β, resulting in the lung metastasis. TGF-β regulates the EMT progression through activation of Smad2,3-4/Snail/E-cadherin signaling pathway. Blockade of TGF-β/TGF-β receptor synapse with TGF-β receptor inhibitor suppresses colorectal cancer cells lung metastasis.
Materials and Methods
1. Patient samples
Colorectal cancer samples were sterilely obtained after the surgery at the first affiliated hospital of Wenzhou Medical University and were sent to the laboratory within 2 hours. According to the clinical data, samples were divided into metastasis and non-metastasis groups. The clinical information was listed in Table 1. All samples were confirmed as colorectal cancer by a pathologist expert.
2. Cell lines and reagents
HCT116 (human colorectal cancer cell line) and CT26 (mouse colorectal cancer cell line) cells were purchased from the Cell Bank of Chinese Academy of Sciences (Shanghai, China). All cell lines were cultured in RPMI-1640 (Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY), penicillin (100 U/mL), and streptomycin (0.1 mg/mL). Recombinant interleukin (IL)-10, TGF-β1, CCL21, CCL18, CXCL12, and vascular endothelial growth factor (VEGF) proteins were purchased from Yanjing Company (Shanghai, China). TGF-β1 neutralization antibody was purchased from Abcam (Cambridge, UK). Clodronate liposomes were purchased from Yisheng Company (Yu Yao, China). LY2109761 was purchased from Selleck (Houston, TX). Anti-CCL2 and anti-mouse CSF1R (CD115) were purchased from Bio X Cell (Lebanon, NH).
3. TAMs preparation and collection from tumor tissues
For mouse TAMs, BALB/c mice (females, 6-8 weeks old) were purchased from Vital River Laboratory Animal Technology Company (Beijing, China). Mice were implanted subcutaneously with 1×105 CT26 cells into the right flank. Mice were scarified and separated the tumor tissues when the tumor reached to 7 mm×7 mm. After digesting the tissues by using collagenase II and IV in 37°C for 2 hours, TAMs were obtained by using separating medium (Junrui Techonology, Shenzhen, China) and sorted with Anti-CD45 and Anti-F4/80 Microbeads (Miltenyi Biotec, Cologne, Germany). For human TAMs, single cell from tumor samples were harvested with DNase and collagenase II and IV digestion in 37°C for 2 hours. TAMs were collected by CD45+CD68+ sorting (eBioscience, San Diego, CA).
4. Lung metastasis assay
BALB/c mice were implanted subcutaneously with 1×105 CT26 cells into the right flank. Twenty-five days later, mice were sacrificed and the lungs were harvested to measure lung weight and examine the number of pulmonary nodules [7]. To further measure the metastasis in lung, the sections were also stained with hematoxylin and eosin (H&E) (Solarbio, Beijing, China).
5. Immunohistochemistry
After sacrificing mice, tissues were kept in 4% paraformaldehyde (PFA) overnight, then the samples were processed, embedded in paraffin, and sectioned at 4 μm for further study. To detect the distribution of CD68 or TGF-β in human colorectal cancer tissues, 10 metastatic samples and 10 non-metastatic samples were conducted by immunohistochemistry (IHC). Antigen retrieval was done using citric acid and sodium citrate in a Microwave oven (Media, Guangdong, China). Then the sections were incubated with CD68 (1:500, Abcam) or TGF-β (1:500, Abcam) at 4°C overnight, followed by signal amplification using a ABC HRP Kit (Thermo Fisher, Waltham, MA) and counter-staining with hematoxylin, dehydration with series of graded ethanol and cleaned with xylene [1]. Microscope (Leica, Barnack, Oskar, Germany) was used to visualize the sections. Ten visions were taken from each sample randomly for intensity analysis and one representative graph were presented.
6. Immunofluorescence
Samples were blocked in 5% bovine serum albumin in phosphate buffered saline (PBS) for 1 hour, E-cadherin (1:200, Abcam) and vimentin (1:300, Abcam) were incubated at 4°C overnight, followed by signal amplification using TSA Kit (PerkinElmer, Waltham, MA) [2]. An Olympus confocal microscope (Tokyo, Japan) was used to visualize.
7. Western blotting
Samples were solubilized with an equal volume of loading buffer (125 mM Tris-HCl, pH 6.8, 4% sodium dodecyl sulfate, 20% glycerol, 0.05% bromophenolblue, 5% β-mercaptoethanol) and were boiled for 10 minutes, then samples were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis, followed by transferring to polyvinylidene difluoride membranes and detecting by immunoblotting with primary antibodies against E-cadherin (1:500, Abcam), vimentin (1:500, Abcam), p-Smad2 (1:500, Abcam), p-Smad3 (1:500, Abcam), Smad4 (1:500, Abcam), and Snail (1:500, Abcam), respectively at 4°C overnight. Then HRP-conjugated secondary antibody (CST, Boston, MA) was incubated for 1 hour at room temperature, and visualized by using ECL detection kit (CST) [3]. β-Actin (1:1,000, Abcam) was used as an internal control.
8. Real-time polymerase chain reaction
The quantification of gene transcripts was performed by real-time polymerase chain reaction (PCR) using SYBR green dye (Solarbio) [4]. Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was used for normalization. The primers used are listed as follows: human GAPDH forward primer 5'-GGAGCGAGATCCCTCCAAAAT-3', reverse primer 5'-GGCTGTTGTCATACTTCTCATGG-3'; human E-cadherin, forward primer 5'-CGAGAGCTACACGTTCACGG-3', reverse primer 5'-GGGTGTCGAGGGAAAAATAGG-3'; human vimentin forward primer 5'-GACG CCATCAACACCGAGTT-3', reverse primer 5'-CTTTGTCGTTGGTTAGCTGGT-3'; human Tgf b1 forward primer 5'- GGCCAGATCCTGTCCAAGC-3', reverse primer 5'-GTGGGTTTCCACCATTAGCAC-3'; human Tgf b receptor forward primer 5'-GTAGCTCTGATGAGTGCAATGAC-3', reverse primer 5'-CAGATATGGCAACTCCAGTG-3'; human IL-10 forward primer 5'-GACTTTAAGGGTTACCTGGGTTG-3', reverse primer 5'-TCACATGCGCCTTGATGTCTG-3'; human IL-10 receptor forward primer 5'-CCTCCGTCTGTGTGGTTTGAA-3', reverse primer 5'-CACTG CGGTAAGGTCATAGGA-3'; human VEGFα forward primer 5'-AGGGCAGAATCATCACGAAGT-3', reverse primer 5'-AGGGTCTCGATTGGATGGCA-3'; human VEFGR forward primer 5'-AGGGCAGAATCATCACGAAGT-3', reverse primer 5'-AGGGTCTCGATTGGATGGCA-3'; human CCL17 forward primer 5'- CGGGACTACCTGGGACCTC-3', reverse primer 5'-CCTCACTGTGGCTCTTCTTCG-3'; human CCR4 forward primer 5'-CCCACGGATATAGCAGACACC-3', reverse primer 5'-GTGCAAGGCTTGGGGATACT -3'; human CCL21 forward primer 5'-GTTGCCTCAAGTACAGCCAAA-3', reverse primer 5'-AGAACAGGATAGCTGGGATGG-3'; human CCR7 forward primer 5'-TGAGGTCACGGACGATTACAT-3', reverse primer 5'-GTAGGCCCACGAAACAAATGAT-3'; human CXCL12 forward primer 5'-ATTCTCAACACTCCAAACTGTGC-3', reverse primer 5'-ACTTTAGCTTCGGGTCAATGC-3'; human CXCR4 forward primer 5'-ACTACACCGAGGAAATGGGCT-3', reverse primer 5'-CCCACAATGCCAGTTAAGAAGA-3'; mouse GAPDH forward primer 5'-AGGTCGGTGTGAACGGATTTG-3', reverse primer 5'-GGGGTCGTTGATGGCAACA-3'; mouse E-cadherin, forward primer 5'-CAGTTCCGAGGTCTACACCTT-3', reverse primer 5'-TGAATCGGGAGTCTTCCGAAAA-3'; mouse vimentin forward primer 5'-CGTCCACACGCACCTACAG-3', reverse primer 5'-GGGGGATGAGGAATAGAGGCT-3'; mouse Tgfb1 forward primer 5'-CCACCTGCAAGACCATCGAC-3', reverse primer 5'-CTGGCGAGCCTTAGTTTGGAC-3'; mouse Tgfb receptor forward primer 5'-AGTCGGATGTGGAAATGGAAG-3', reverse primer 5'-CTGGCCATGACATCACTGTTA-3'; mouse IL-10 forward primer 5'-CTTACTGACTGGCATGAGGATCA-3', reverse primer 5'-GCAGCTCTAGGAGCATGTGG-3'; mouse IL-10 receptor forward primer 5'-TTGTCGCGTTTGCTCCCATT-3', reverse primer 5'-GAAGGGCTTGGCAGTTCTG-3'; mouse VEGFα forward primer 5'-CTGCCGTCCGATTGAGACC-3', reverse primer 5'-CCCCTCCTTGTACCACTGTC-3'; mouse VEGFα receptor forward primer 5'-TTTGGCAAATACAACCCTTCAGA-3', reverse primer 5'-GCTC CAGTATCATTTCCAACCA-3'; mouse CCL17 forward primer 5'-TACCATGAGGTCACTTCAGATGC-3', reverse primer 5'-GCACTCTCGGCCTACATTGG-3'; mouse CCR4 forward primer 5'-TGCACCAAGGAAGGTATCAAGG-3', reverse primer 5'-GTACACGTCCGTCATGGACTT-3'; mouse CCL21 forward primer 5'-GTGATGGAGGGGGTCAGGA-3', reverse primer 5'-GGGATGGGACAGCCTAAACT-3'; mouse CCR7 forward primer 5'-CAGGTGTGCTTCTGCCAAGAT-3', reverse primer 5'-GGTAGGTATCCGTCATGGTCT-3'; mouse CXCL12 forward primer 5'-TGCATCAGTGACGGTAAACCA-3', reverse primer 5'-CACAGTTTGGAGTGTTGAGGAT-3'; mouse CXCR4 forward primer 5'-GACTGGCATAGTCGGCAATG-3', reverse primer 5'-AGAAGGGGAGTGTGATGACAAA-3'.
9. Cell invasion assay
For invasion assay, 30 μg matrigel (BD Biosciences, FranklinLakes, NJ) was used to coat the upper chamber of 24 wells plates (8 μm, Corning Company, Corning, NY). Then, CT26 or HCT116 cells (3×104) suspended in fetal calf serum (FCS)‒free 1640 RPMI medium were seeded in the upper chamber, meanwhile, the lower chamber was added with macrophage culturing supernatants. RPMI-1640 without FCS was used as the negative control. Cells were allowed to migrate at 37°C for 18 hours. Then 4% PFA was used to fix the cells that invaded the lower surface of the membrane, followed by 1% crystal violet staining for calculation [5]. In some experiments, the lower chamber was added with the following recombinant proteins, IL-10/TGF-β/CCL21/CCL18/CXCL12/VEGF, to determining whether they involve in the cell invasion process.
10. Wound healing assay
For wound healing assay, CT26 or HCT116 cells were seeded in 6-well plates, a 10-μL microtiter tip was used to cut trace longitudinally when cells reaching 85% confluent, then cells were treated with or without macrophage culturing supernatants or TGF-β for 24 hours [6]. Using the microscope, pictures were taken as 200× for analysis.
11. RNA interfering
For Snail knockdown in tumor cells, 2×105 CT26 cells/well were seeded in a six-well plate and starved in an antibioticfree growth medium for 24 hours before transfection. Snail siRNA (4 mL, 0.5 mg) or a mock siRNA solution was performed for 48 hours according to the manufacturer’s protocol. The transfection efficacy was validated by western blot [9]. The primers were designed and constructed by Gene Pharma Company (South San Francisco, CA). Then the cells were treated with TGF-β to examine the cell morphology in microscope. The following sequences were successfully: siRNA-Snail sense 5'-GAT CCG CCT AAC TAC AGC GAG CTG TTC AAG AGA CAG CTC GCT GTA GTT AGG CTT TTT TGG AAA-3' and antisense 5'-AGC TTT TCC AAA AAA GCC TAA CTA CAG CGA GCT GTC TCT TGA ACA GCT CGC TGT AGT TAG GCG-3'; the mock vector sense 5'-GAT CCG TAT TGC CTA GCA TTA CGT TTC AAG AGA ACG TAA TGC TAG GCA ATA CTT TTT TGG AAA-3' and antisense 5'-AGC TTT TCC AAA GTA TTG CCT AGC ATT ACG TTC TCT TGA AAC GTA ATG CTA GGC AAT ACG-3'.
12. Flow cytometry
To examine the percentage of macrophage in patients, cells were incubated with CD68 (eBioscience) for 20 minutes at room temperature, after washed twice and then re-suspended in PBS [10]. For intracellular TGF-β (eBioscience) analysis, mice TAMs and peritoneal macrophages or human TAMs and peripheral blood monocytes were collected and fixed, after permeabilization, anti-human or anti-mouse TGF-β were stained respectively. Flow cytometry was performed on the BD Canto II (BD Biosciences). 7-AAD was used to exclude the dead cells. IgG (Abcam) was used as the negative control.
13. Animal protocols and treatments
To evaluate the effects of TGF-β on metastasis in colorectal cancer, 5×105 CT26 were subcutaneous injected into the BALB/c mice (female, 6-8 weeks, Huafukang, Shenzheng, China). On day 15, mice were treated with PBS, TGF-β (250 ng), TAMs cultural supernatant and TAMs cultural supernatant combined with TGF-β neutralizing antibody (200 μg) by intratumoral injection. Mice were sacrificed for the lung metastasis analysis on day 25. To evaluate the anti-metastasis effects of LY2109761, 5×105 CT26 were subcutaneous injected into the BALB/c mice. On day 15, 18, and 21, mice were treated with PBS or LY2109761 (50 mg/kg) by intratumoral injection. On day 25, the mice were sacrificed for the lung metastasis analysis.
14. Enzyme-linked immunosorbent assay
The concentrations of TGF-β in tumor tissues from metastatic and non-metastatic patients were quantified by using commercially available enzyme-linked immunosorbent assay (ELISA) Kits (Dakewe Biotech Co., Ltd., Shenzhen, China). Assays were performed according to the manufacturer’s protocol and read at 450 nm by using a microplate reader (Thermo Scientific).
Results
1. TAMs facilitate colorectal cancer cells metastasis
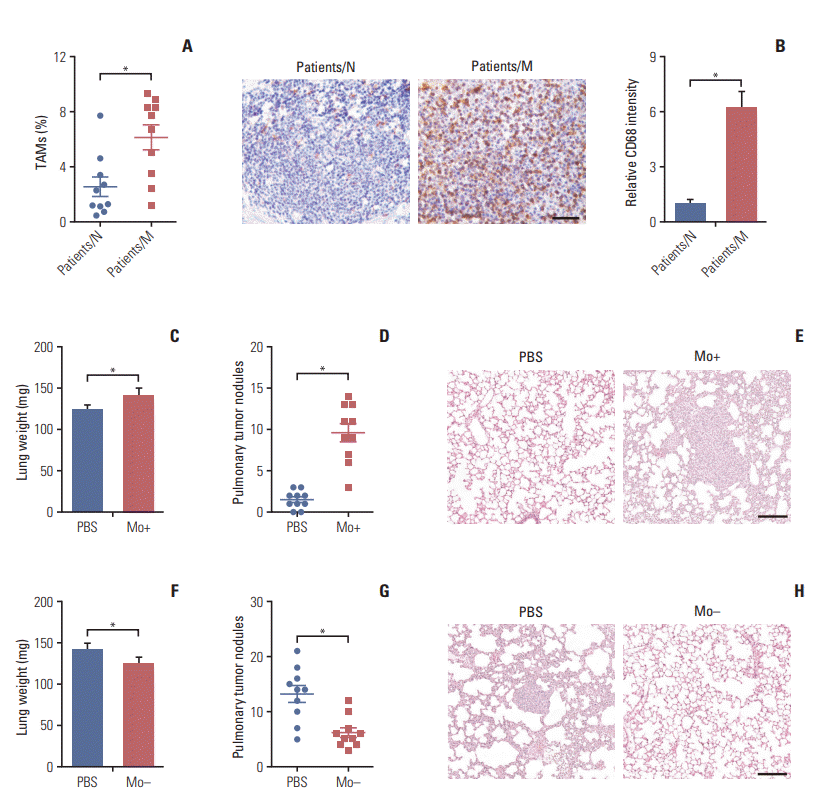
Recently, TAMs accumulation level has been shown to be of significant in cancer cells metastasis in a variety of cancers. To test the infiltration level of TAMs in colorectal cancer, we analyzed CD68+ cells in CD45+ cells in metastatic (patients/M) and non-metastatic (patients/N) colorectal cancer samples from clinical patients by flow cytometry. Interestingly, we found that metastatic patients have more TAMs accumulated than non-metastatic patients in tumor tissues (Fig. 1A). Moreover, we verified this by IHC and the CD68 IHC intensity was correlated with flow cytometry analysis (Fig. 1B). It has been reported that the accumulated TAMs in breast cancer and lung cancer could induce the metastasis. Our initial results suggested that colorectal cancer tissues have TAMs accumulation and the infiltration level was much higher in metastatic patients’ tissues than non-metastatic patients’ (Fig. 1A and B). To clarify whether TAMs could promote the colorectal cancer metastasis, the CT26 mouse subcutaneous model was established. We injected 105 macrophages which was isolated from tumor tissues into tumor site. On the day 25, mice were sacrificed, lungs were weighed, and the metastatic pulmonary tumor nodules on the surface of lungs were counted. We found that the macrophages injection increased the lung weight compared with PBS injection group (Fig. 1C) and increased the pulmonary tumor nodules in lung as well (Fig. 1D). Further, we observed that CT26 cells seeded more lung metastasis with macrophages injection, which was confirmed by the whole lung H&E staining (Fig. 1E). These results indicate that TAMs could promote the colorectal cancer cells lung metastatic seeding.
To further determine whether TAMs were involved in colorectal cancer lung metastasis, we used clodronate liposomes to deplete the macrophages in tumor-bearing mice. We found that macrophages depletion significantly reduced the colorectal cancer cells lung metastasis, which was supported that the macrophages depletion group has the lighter lung weight (Fig. 1F), fewer pulmonary tumor nodules (Fig. 1G) and less lung metastasis (Fig. 1H) than PBS group. Moreover, we used anti-CCL2 and anti-CSF1R (CD115) to target macrophages in CT26-bearing mice, both the lung weight and pulmonary tumor nodules were reduced (S1A and S1B Fig.). Together, our data suggest that TAMs could promote colorectal cancer lung metastasis.
2. TAMs promote colorectal cancer cells metastasis via the secretion of TGF-β
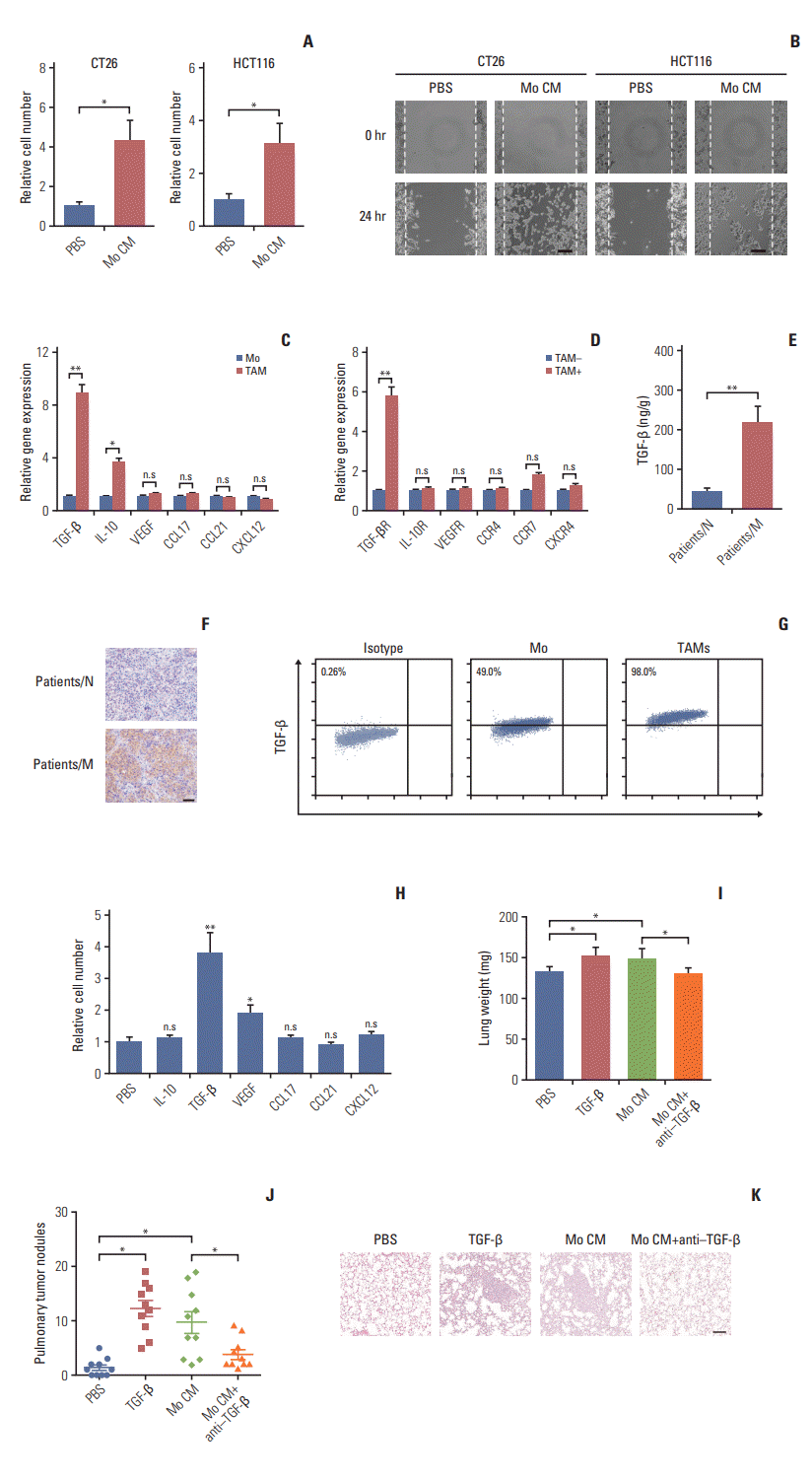
Our previous data showed that TAMs was involved in colorectal cancer cells lung metastasis. Next, we were wondering that how TAMs promote colorectal cancer cells metastasis. On one hand, we performed the transwell experiments and found that the invasion cells were significantly increased in CT26 and HCT116 with TAMs conditioned medium treatment (Fig. 2A). On the other hand, the cell migration was enhanced in TAMs conditioned medium treated CT26 and HCT116 by wound healing assay (Fig. 2B). Those data indicate that TAMs may secret some kind of protein to induce colorectal cancer cells invasion and migration.
Several enriched protein secreted by macrophages in tumor microenvironment, like IL-10, TGF-β, VEGF, CCL18, CCL21, CXCL12 and so on, have been reported as the functional factors that responsible for the tumor progress [21]. To identify which secreted factors drive colorectal cancer cells invasion, we used real-time PCR to screen those molecules in normal macrophages or TAMs and we found that only TGF-β was increased in both mice (Fig. 2C) and human TAMs (S2A Fig.). Further, we detected the corresponding receptors in CT26 and HCT116 cells, TGF-β receptor was upregulated with TAMs medium treatment (Fig. 2D, S2B Fig.). Those data suggest that TGF-β/TGF-β receptor synapse may be involved in colorectal cancer cells invasion and migration. To better demonstrate TGF-β was produced from TAMs to mediate the above cell activity, firstly, we measured the TGF-β level in metastatic patients and non-metastatic patients’ tissues by ELISA and IHC, data showed that metastatic patients’ tumor tissues have more TGF-β accumulated (Fig. 2E and F). Moreover, we found almost all mice and human TAMs could produce TGF-β and TAMs possessed more TGF-β than peritoneal macrophages or peripheral blood monocytes (Fig. 2G, S2C Fig.). Next, the recombinant proteins were used to treat CT26 and HCT116 cells in transwell experiments. Predictably, only recombinant TGF-β significantly increased the cell invasion (Fig. 2H, S2D Fig.). Consistently, colorectal TAMs could promote cancer cells invasion and migration in terms of TGF-β. Taking together, these data suggest that TGF-β may be the main factor that responsible for the colorectal cancer metastasis induced by TAMs.
Furthermore, to confirm TGF-β induced colorectal cancer lung metastasis in vivo, we injected conditioned medium from TAMs and recombinant TGF-β into the CT26-bearing mice. We found that conditioned medium from TAMs and recombinant TGF-β significantly increased lung weight (Fig. 2I), the pulmonary tumor nodules (Fig. 2J) and lung metastasis (Fig. 2K) comparing with PBS group. However, adding anti–TGF-β neutralizing antibody could reverse those effects (Fig. 2I-K). Collectively, these results indicate that TAMs could secret TGF-β to promote colorectal cancer cells lung metastatic seeding.
3. TGF-β induces the colorectal cancer cells metastasis through the EMT progression
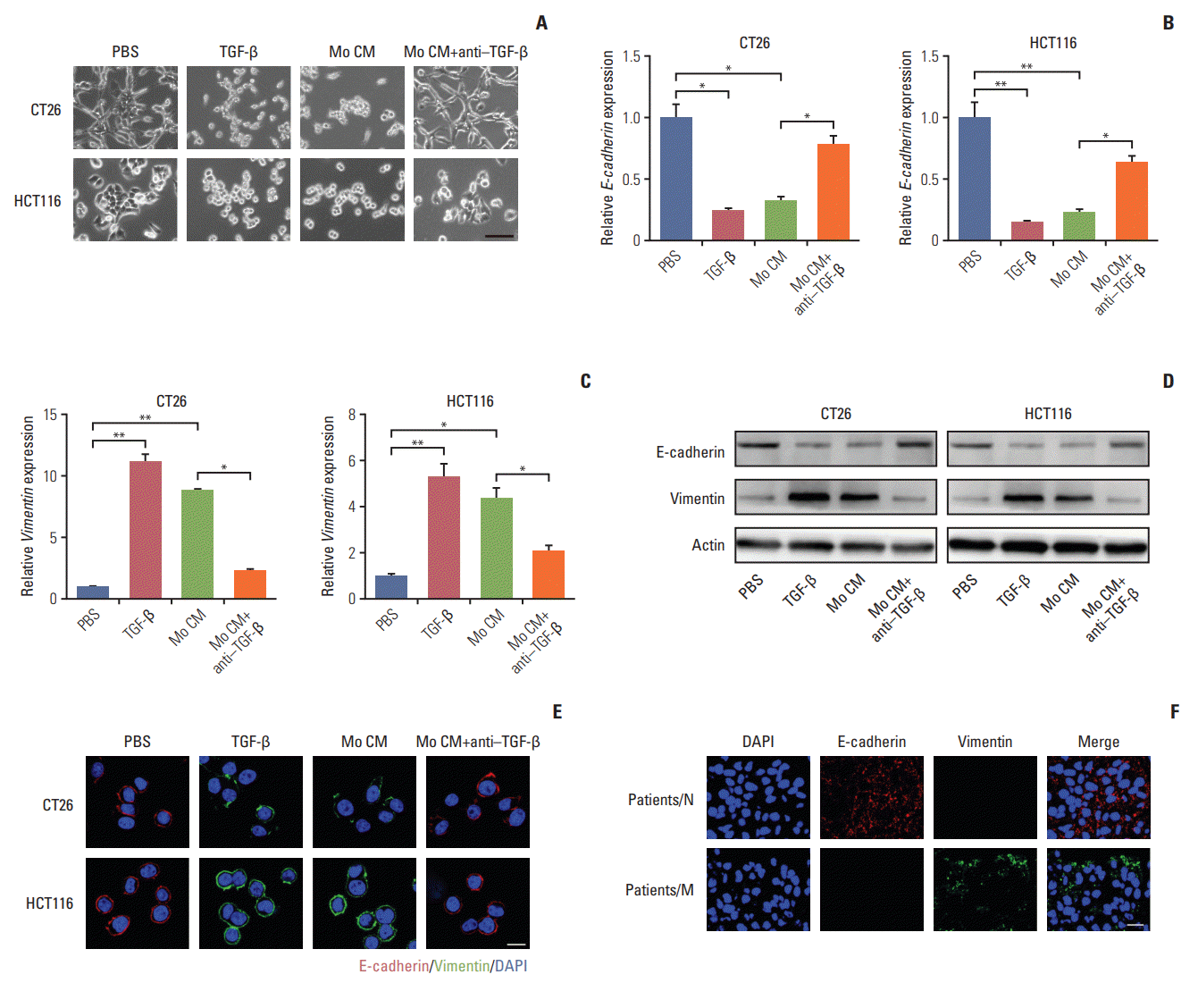
We have found that TGF-β could induce colorectal cancer cells invasion and migration and it has been verified that TGF-β could initiate the EMT to enhance tumor cell invasion and migration in a variety of cancers, which was characterized as loss of E-cadherin and elevated vimentin expression [22-24]. We were wondering that whether TGF-β could trigger EMT in colorectal cancer cells. In order to visually capture the morphological change of EMT in vitro, we conducted the cell imaging of CT26 and HCT116 cells with TGF-β or conditioned medium from TAMs treatment. We observed that treating with TGF-β and conditioned medium from TAMs could significantly induce epithelioid CT26 and HCT-116 cells transition into mesenchymal phenotype (Fig. 3A), while adding anti–TGF-β antibody reversed the effect (Fig. 3A). Moreover, E-cadherin, a hallmark of epithelial cells, was significantly decreased with TGF-β and conditioned medium from TAMs treatment in CT26 and HCT116 cells (Fig. 3B, D, and E). Concomitantly, vimentin, one of the mesenchymal cytoskeletal proteins, was remarkably increased with TGF-β and conditioned medium from TAMs treatment (Fig. 3C-E). Conversely, adding anti‒TGF-β antibody effectively reversed the effect (Fig. 3B-E). These data suggest that TGF-β could induce the colorectal cancer cells EMT. Accumulating evidence supported that EMT was correlated with the metastasis and poor prognosis in cancer patients [25,26]. Here, to confirm colorectal cancer cells metastasis was EMT involved, we analyzed the expression of EMT markers in cancer patients’ tissues. We found that vimentin mainly expressed in metastatic patients’ tissue, while non-metastatic patients’ tissue had plenty E-cadherin expression (Fig. 3F). This clinical data demonstrated that EMT was involved in colorectal cancer cells metastasis. Taking together, those results conclude that TGF-β induced EMT is involved in colorectal cancer cells metastasis.
4. TGF-β mediates colorectal cancer cells EMT via Smad2, 3-4/Snail/E-cadherin pathway
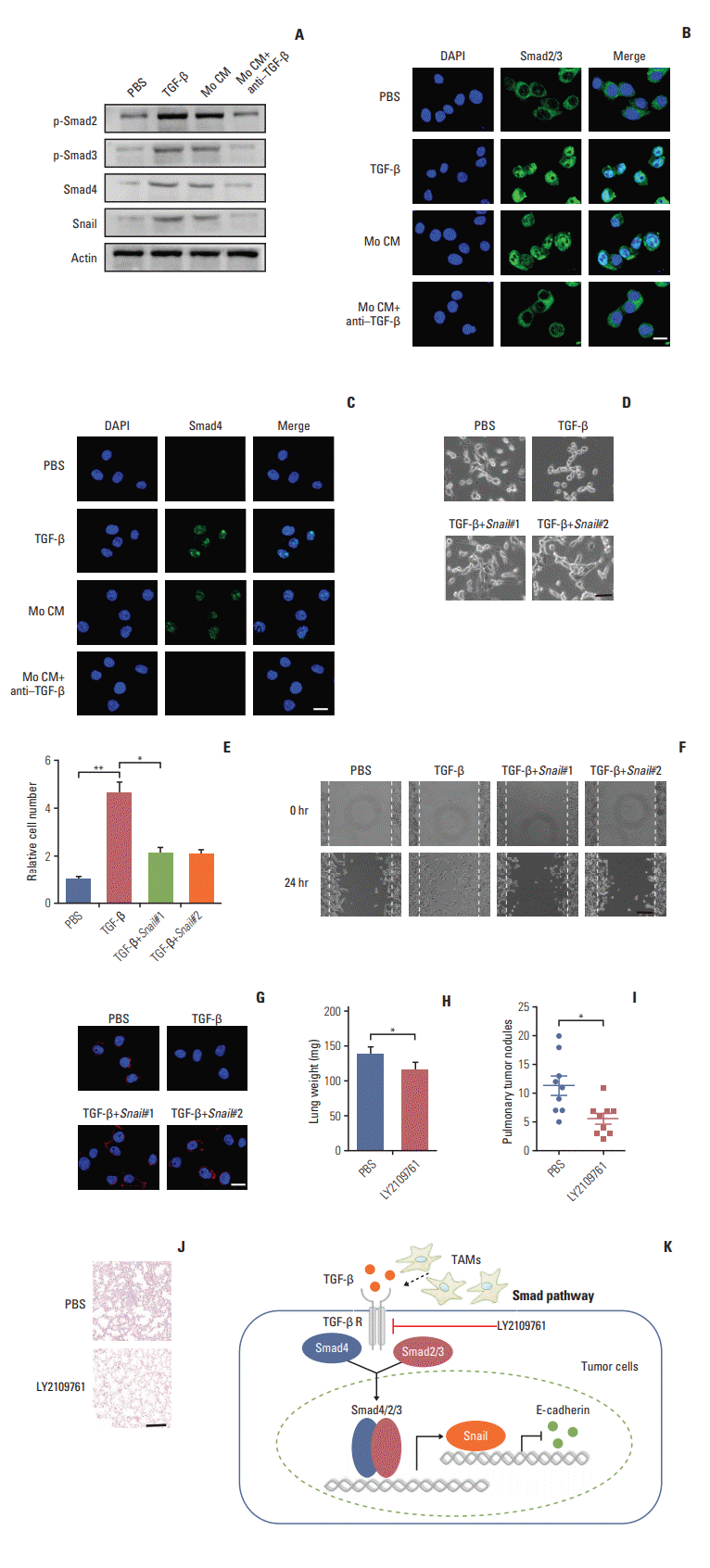
Next, we want to figure out how TGF-β endowed the EMT of colorectal cancer cells and to explore potential therapeutic target in clinic. The signaling mechanisms of TGF-β induced EMT has been largely conducted in a variety of cancer cells. In response to TGF-β, the cellular Smad signaling would be activated by phosphorylated TGF-β receptors [27]. To test whether the Smad signaling involved in the colorectal cancer cells EMT, we detected the activation of Smads with TGF-β or conditioned medium from TAMs in CT26 colorectal cancer cells. We found that TGF-β or conditioned medium from TAMs increased the phosphorylation of Smad2/3, and significantly enhanced the expression of Smad4 (Fig. 4A). Also, we observed that the Smad2/3 and Smad4 complex was translocated into nucleus (Fig. 4B and C). Moreover, Snail, the downstream transcription factor of Smad, was increased by TGF-β and conditioned medium from TAMs treatment (Fig. 4A). While pre-treatment with TGF-β neutralizing antibody, reversed the effect (Fig. 4A-C). Further, siRNA targeting Snail transcripts suppressed the epithelia to mesenchymal phenotype changes in TGF-β treated CT26 cells (Fig. 4D). Furthermore, TGF-β induced colorectal cancer cells invasion and migration were significantly inhibited with Snail knockdown (Fig. 4E and F). In line with the pervious results, TFG-β suppressed the expression of E-cadherin. Here, the silence of Snail could efficiently reverse the effects (Fig. 4G). Those data suggest that TGF-β endowed colorectal cancer cells EMT was Snail dependent. In order to disrupt the TAMs triggered colorectal cancer cells metastasis in vivo, we used TGF-β receptor inhibitor to treat CT26-bearing mice. Exhilaratingly, the lung metastasis was remarkably suppressed in LY21-09761 group compared with PBS group with lighter lung weight and less pulmonary tumor nodules (Fig. 4H-J). Collectively, these results indicate that TGF-β activates the Smads/Snail signaling pathway to induce colorectal cancer cells EMT for metastasis.
Discussion
Cumulated evidences support that TAMs involved in tumor cell metastasis, which is mediated through the secretion of cytokines [28]. Recently, TAMs have been reported to accumulate in the tumor sites and promoted distant metastasis in various cancers [28]. Further studies are required to reveal the mechanism of TAMs and determine whether could improve the clinical efficiency by blocking pathway.
In our study, we found that TAMs isolated from primary tumor tissues promote colorectal cancer cells invasion and migration in vitro and in vivo which provided evidence to support the concept that TAMs play a crucial role in colorectal cancer metastasis. It was consistent with previous research revealing that accumulated TAMs in tumor microenvironment participate in different tumor progression, including cancer stemness maintain, tumor growth and drug resistance, through the secretion of cytokines. Tumor distant metastasis is considered as the main cause of cancer-related deaths, and EMT is regarded as the key steps. Further, our study indicated that TAMs could facilitate the colorectal cancer metastasis through inducing EMT of tumor cells by secretion of TGF-βwhich was validated by in vitro and in vivo data [29]. Also, our results showed that TGF-β produced by TAMs in tumor microenvironments activated the Smad signaling pathway by binding to the TGF-β receptors, followed by the phosphorylated Smad2/Smad3 to form complex with Smad4 and regulate transcription of Snail. Once the TGF-β triggered the colorectal cancer cells EMT, Snail could repress the expression of epithelial marker E-cadherin, resulting in the cancer metastasis (Fig. 4K).
Various crucial features are believed to participate in the cancer metastasis progress, including the expression of integrin [30], tumor cells EMT progression, tumor microenvironments [11]. It has been demonstrated that TAMs play a crucial role in different stages of tumor progression [31]. TAMs facilitate the sustained growth of tumor cells through secretion of growth factors such as CCL8 [20] and remodeling the tumor microenvironments by matrix metalloproteinases (MMPs). Moreover, TAMs promote tumor cells invasion and migration through the secretion of tumor necrosis factor and MMPs [19]. However, the specific mechanism of metastasis induced by TAMs is still unclear. In our studies, we further demonstrate that TAMs could facilitate the metastasis through the secretion of TFG-β in colorectal cancer cells.
Increasing evidences demonstrated that tumor cells could promote metastasis through an autocrine manner. Various cancer cells, including breast cancer cells and pancreatic cancer cells, undergo the progression of EMT before metastasis [29], which is induced by the TGF-β produced by the tumor cells. Our data found that TGF-β could be derived from TAMs and suggested that inhibition of TGF-β signaling significantly decreased the EMT. And the specific signaling pathways of EMT in several cancer cells remained to be unclear. We investigated the progression of EMT in colorectal cancer and found that a Smad dependent signaling pathway, which is induced by the TGF-β produced from TAMs in tumor microenvironment. The data supports TAMs endowed with the capacity to facilitate the metastasis and regulate the tumor progression, readjusting the focus of inhibit tumor growth to selectively suppress TAMs functions and remodeling of tumor microenvironment should be considered when targeting colorectal cancer metastasis in clinic.
In summary, our data provided evidence to support that accumulated TAMs in colorectal cancers contributed to distant metastasis through secreting TGF-βwhich induced EMT by activating Smad2,3-4/Snail pathway. Blocking TGF-β signaling remarkably reduced the EMT which in turn resulted in decreased metastasis. Our data laid an important foundation for potential application of TGF-β inhibition in clinical treatment as anti-metastatic therapy for colorectal cancer patients.
 XML Download
XML Download