

 PDF
PDF Citation
Citation Print
Print


Introduction
Homeobox (HOX) genes are essential developmental regulators that control a wide range of processes, including apoptosis, differentiation, motility, and angiogenesis [1]. In humans, there are four HOX clusters (A, B, C, and D), and 39 HOX genes have been identified [1]. The HOX genes are normally active during embryogenesis, but most are not expressed or are expressed at very low levels in an adult brain [2]. On the other hand, several studies have indicated aberrant expression of the HOX genes in brain tumors as well in as other cancers from various organs [3-6]. Moreover, there is growing clinical evidence of a prognostic effect of HOX gene expression in several cancers [7-10].
Aberrantly expressed HOX genes in cancer cells have multicapacity functions, including metastasis, tumor growth, anti-apoptosis, and differentiation suppression [1]. The overexpression of multiple HOX genes have been observed in glioblastoma (GBM) cell lines and primary astrocytoma [4]. In addition, some studies have shown that the HOX genes are important in the treatment resistance of GBM [11-13]. On the other hand, the precise mechanism showing the role of HOX genes and their functional relevance in glioma cells is unclear. GBMs, similar to other cancers, harbor a cell subpopulation with a stem cell-like capacity that is associated with the development of a tumor progeny and treatment resistance [9,14,15]. Given the roles of HOX genes in development and organogenesis, it has been postulated that a portion of the relative expression of HOX genes is integral to stem cell activity, specifically self-renewal, tissue specificity, and quiescence [1]. Among the HOX genes, the HOXA cluster is important in human embryonic stem cell differentiation [16]. Collectively, these results suggest that HOX genes are plausible candidates as biomarkers for assessing the prognosis of GBM and a credible target for overcoming the treatment resistance in GBM.
HOX genes were previously reported to be the genes of interest related to GBM recurrence and treatment resistance [17]. Moreover, a mechanism through which the HOXA10 gene affects temozolomide (TMZ) resistance in GBM cell lines was reported [12]. HOXA10 induces the transcription of early growth response 1, which results in phosphatase and tensin homolong (PTEN) and Rad51 paralogs. As a result, the homologous recombination DNA repair system with Rad51 genes can protect cancer cells from TMZ-induced cytotoxicity [12].
In the present study, this research was extended to another HOX gene, HOXA11, and its role in GBM prognosis was examined.
Materials and Methods
1. Patient samples and cell lines
Fresh frozen tumor tissue samples of five GBM patients, in whom of pairs of initial and recurrent samples were available for screening, and another 20 newly diagnosed GBM patients for validation were used in this study. All patients were managed using a standard GBM treatment protocol of concurrent radiotherapy and TMZ treatment, followed by adjuvant TMZ as a primary treatment. The tumor tissues were obtained during surgery, snap-frozen in liquid nitrogen, and stored at −80°C prior to use. The study was approved by an institutional review committee.
The human glioma U251, U373, and LN18 cell lines were purchased from the American Type Culture Collection (Manassas, VA), and cultured in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and 5% antibiotics (streptomycin) in a humidified atmosphere containing 5% CO2 and 95% air at 37°C.
2. Reverse transcription polymerase chain reaction
The cell lines were lysed with TRIzol (Life Technologies, Carlsbad, CA), and RNA isolation was performed using an RNeasy Mini Kit (#74104, Qiagen, Valencia, CA). The total RNA was treated with DNase and then quantified by spectrophotometry. In addition, cDNA was synthesized from 1 μg of the total RNA using a reverse transcription kit (#205311, Qiagen) according to the manufacturer’s protocol. The primers used were designed using an online primer-BLAST tool (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). The primer sequences for HOXA11 were 5'-GATTTCTCCAGCCTCCCTTC-3' (forward) and 5'-AGAAATTGGACGAGACTGCG-3' (reverse). Using these primers, a reverse transcription polymerase chain reaction (RT-PCR) was performed for 35 cycles. Each cycle was comprised of 95°C for 30 seconds, 62°C for 30 seconds, and 72°C for 45 seconds with each primer set. The RT-PCR products were resolved by 2% agarose gel electrophoresis.
3. Western blot
The whole protein extracts from the tissue samples were prepared using a PRO-PREP lysis buffer (#17081, iNtRon Biotechnology, Seongnam, Korea), and the protein concentrations were determined using a bicinchoninic acid protein assay (#23227, Thermo Fisher Scientific, Waltham, MA). The proteins were separated using 10% sodium dodecyl sulfate polyacrylamide gel electrophoresis, followed by blotting onto nitrocellulose membranes, and probing with the antibodies against HOXA11 (#SC-48542, 1:500 dilution, Santa Cruz Biotechnology, Santa Cruz, CA). The blotted membranes were then incubated with the goat anti-rabbit IgG secondary antibody for 1 hour. Subsequently, the membranes were incubated in an Amersham ECL-prime solution (#RPN2232, GE Healthcare Life Sciences, Pittsburgh, PA) in the dark for 1 minute and exposed under FluorChemHD2 (Cell Biosciences, Santa Clara, CA) for visualization. Only the samples with a consistent result from repeated experiments were selected for analysis. The densities of the bands were measured using free image analyzer software (ImageJ V1.8x, National Institutes of Health, http://rsb.info.nih.gov/ij/). The results are presented as the mean±standard error of mean calculated from independent samples.
4. RNA interference
To knock down HOXA11 expression in cells, small interfering RNA (siRNA) experiments were performed using commercially available sequences targeting HOXA11 (#SASI-Hs01-00110410, #SASI-Hs01-00110413, and #SASI-Hs01-00110417, Sigma Aldrich, St. Louis, MO) as well as with the non-targeting control siRNA (#D-001610-01-05, Dharmacon, Lafayette, CO). When the cells reached 70%-80% confluence, they were transfected with siRNA under the most efficient transfection condition, as determined by the NEON Transfection system (#MPK5000, Life Technologies). The cells were cultured in media without antibiotics to increase the siRNA transfection efficiency for 24 hours.
5. Cell viability analysis after drug and radiation treatment
The control and transfected cells were grown on 96-well plates at a density of 4×103 cells per well for 24 hours. Subsequently, the cells were either treated with TMZ (#ALX-420-044-M100, Enzo Life Sciences, Farmingdale, NY) to a final concentration of 300 μg/mL for 24 hours or irradiated with 4 MV X-rays from a linear accelerator (Clinac 4/100, Varian Medical Systems, Palo Alto, CA) at a dose rate of 10.0 Gy/min. For the combination treatment, the cells were irradiated first and then treated with TMZ.
Cell viability analysis was performed using a Colorimetric Cell Counting Kit-8 (CCK; Dojindo Molecular Technologies, Kumamoto, Japan). The number of viable cells was quantified according to the manufacturer’s instructions by reading the ultraviolet absorption spectra at 450 nm on a microplate 2 hours after adding 10 μL of a CCK solution per well. All experiments were conducted in triplicate.
6. Microarray
For tissue sample analysis, the total RNA was extracted from the tissue samples using the mirVana miRNA Isolation Kit (#AM1560, Ambion, Austin, TX) for microarray analysis after quantification and qualification. The total RNA quality was determined using an Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The cut off RNA integrity number for RNA used in RNA amplification was 7.0 or above. The cRNA was produced using an Illumina TotalPrep RNA Amplification Kit (#IL1791, Ambion) according to the provided protocol. The cRNA was used for hybridization to a human HT12-v4 Illumina Beadchip gene expression array (Illumina, San Diego, CA) according to the manufacturer’s protocol. The arrays were scanned and the fluorescence signals were obtained using an Illumina BeadArray Reader (BeadStation 500GXDW, Illumina). The signal obtained from the scanned beadchip was transformed to intensity raw data using GenomeSortudio software (ver. 2009.1, Illumina) and was used for further data analysis. The raw data were normalized by applying a log2 transformation, quantile normalization, and gene and array centering. All data processing was performed using the R/Bioconductor packages (ver. 2.14, http://www.bioconductor.org).
To determine the changes in gene expression before and after HOXA11 knockdown, the total RNA extracted from the LN18 cells transduced with siHOXA11 or control siRNA were analyzed by Affymetrix GeneChip Human Gene 1.0ST Arrays (Affymetrix, Santa Clara, CA). The RNA was amplified and labeled using a GeneChip WT Sense Target Labeling and Control Reagents Kit (Affymetrix). The cDNA was synthesized, labeled, and hybridized to the GeneChip array according to the manufacturer's protocol. The GeneChips were washed and stained using the GeneChip Fluidics Station 450 (Affymetrix), and then scanned using a GeneChip Scanner 3000 7G (Affymetrix). The expression data were normalized using the robust multi-array average method. Affymetrix Expression Console ver. 1.1 (Affymetrix) was used to compare the group signals, and the data were logtransformed (base 2) for parametric analysis. The differentially expressed genes (DEGs) were identified by significance analysis of the microarrays method in the R package ‘samr’ (R 2.11.1).
7. Statistical analysis
The data from the experiments were tested for their significance using an unpaired two-tailed Student's t test. An ANOVA and Student's t test were used to identify the significant differences in cell death rates. Kaplan-Meier curve analysis was used to generate the overall survival curves. The differences between the survival curves were analyzed using a log-rank test. The results were analyzed using IBM SPSS Statistics software ver. 19.0 (IBM Co., Armonk, NY). The data are presented as mean±standard deviation for three or more separate experiments. A p-value of 0.05 or lower was considered significant.
For microarray analyses, the false discovery rates (FDRs) were calculated using three GenePattern software modules (http://www.broadinstitute.org/cancer/software/genepattern; ComparativeMarkerSelection ver. 10, HierarchicalClustering ver. 6, and HeatMapViewer ver. 13) [18]. The cutoff value for FDR significance was < 0.05. The significantly regulated genes were subjected to functional gene classification using the DAVID Bioinformatics Resources annotation tool (ver. 6.7, http://david.abcc.ncifcrf.gov/) [19]. The selected gene IDs of the identified DEGs were entered into GeneMANIA software (ver. 3.1.2.8, http://www.genemania.org) for network analysis [20].
Results
1. Down-regulation of HOXA11 is associated with a poor prognosis in GBM patients
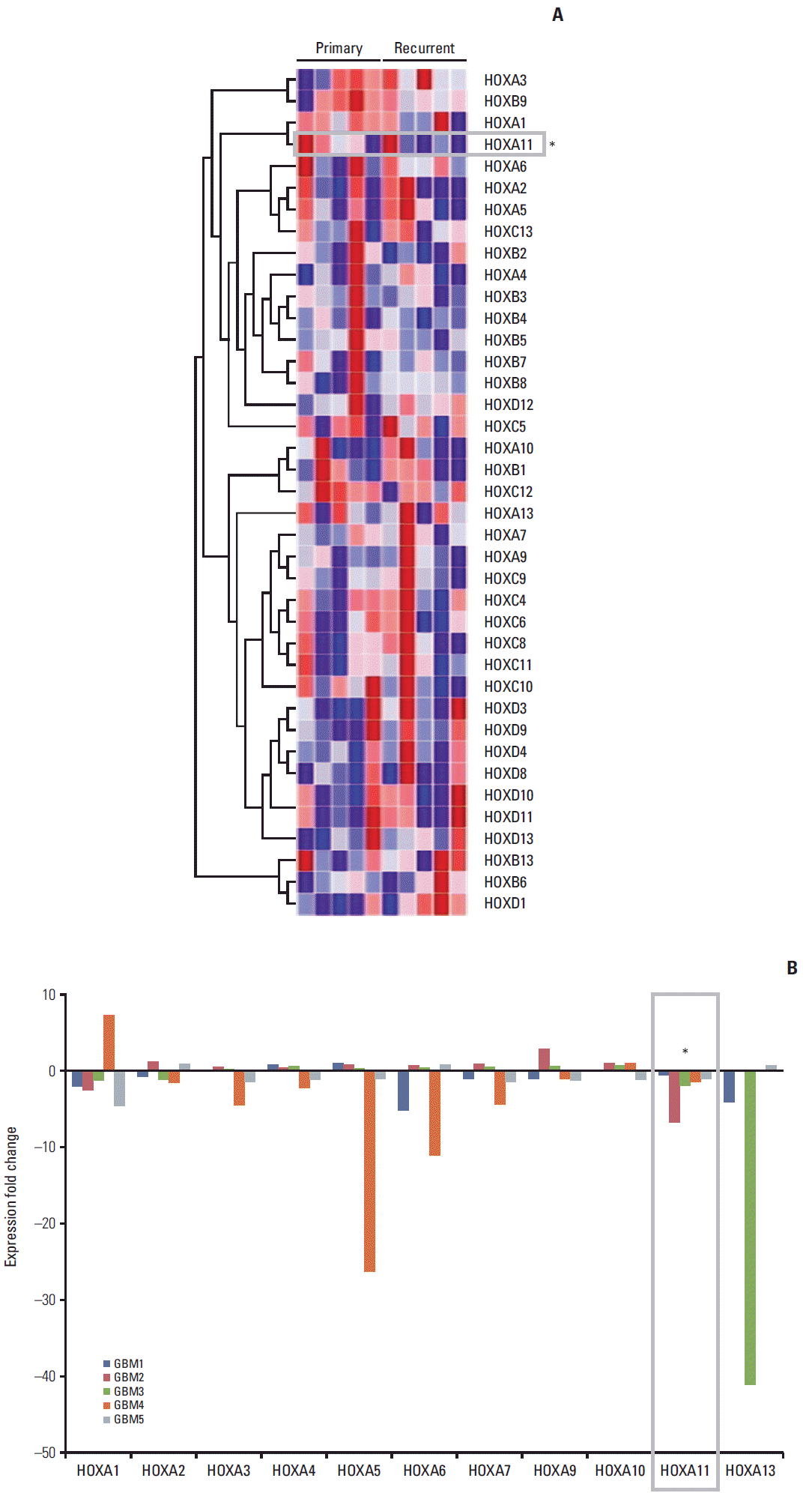
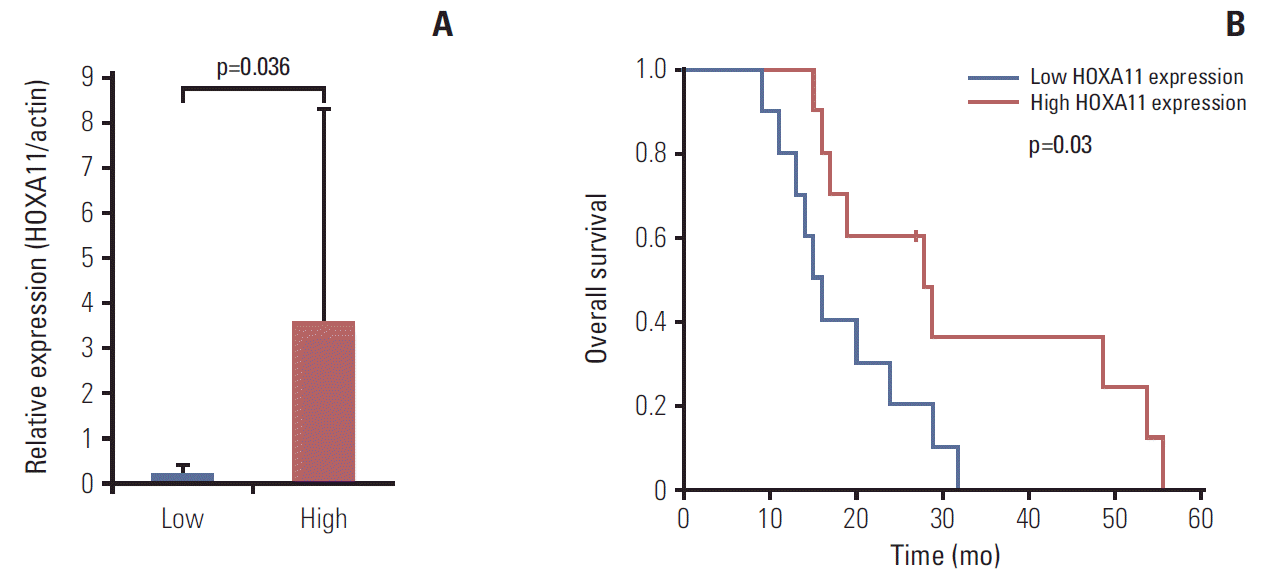
The relative changes in HOX gene expression in five pairs of primary and recurrent GBM samples were assessed using microarray analysis (Fig. 1). Among the 39 HOX genes, HOXA11 was the only gene that consistently showed a significant down-regulation in the recurrent samples (p=0.046). The overall survival of a separate set of 20 GBM patients indicated significantly favorable prognoses in those with high HOXA11 expression compared to those with low HOXA11 protein expression (survival, 31±15.3 months with high HOXA11 expression vs. 18±7.3 months with low HOXA11 expression, p=0.03; expression status determined by western blot analysis) (Fig. 2). Therefore, the down-regulation of HOXA11 is associated with a poor prognosis in GBM patients.
2. Suppression of HOXA11 mediates the treatment resistance in vitro
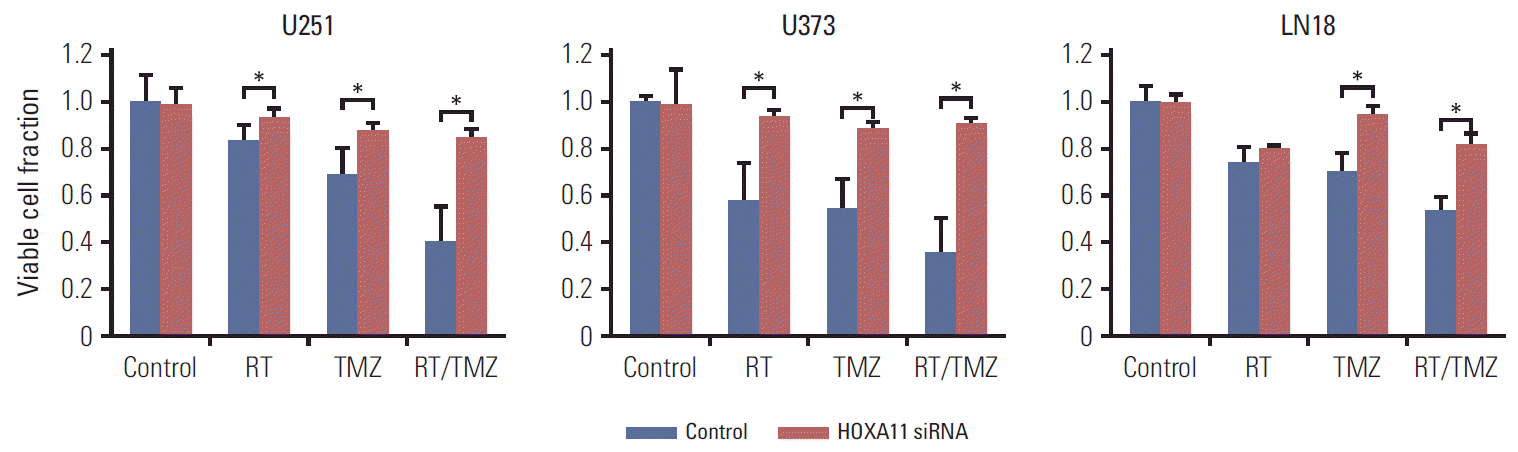
To confirm the effects of HOXA11 expression on the resistance to the current standard treatment protocol for GBM, three malignant glioma cell lines (U251, U373, and LN18) were transduced with HOXA11 siRNA to assess the cell viability. The viable cell fractions were analyzed 72 hours after treatment with either single or combination applications of radiation treatment (RT) and TMZ. A significant increase in the number of cells with HOXA11 suppression after either RT or TMZ was observed (Table 1, Fig. 3). When HOXA11 was suppressed, the treatment resistance effect was more pronounced after treatment with a combination of RT and TMZ (p=0.022 for RT vs. RT/TMZ and p=0.053 for TMZ vs. RT/TMZ).
3. Mediators of HOXA11-related oncologic effect
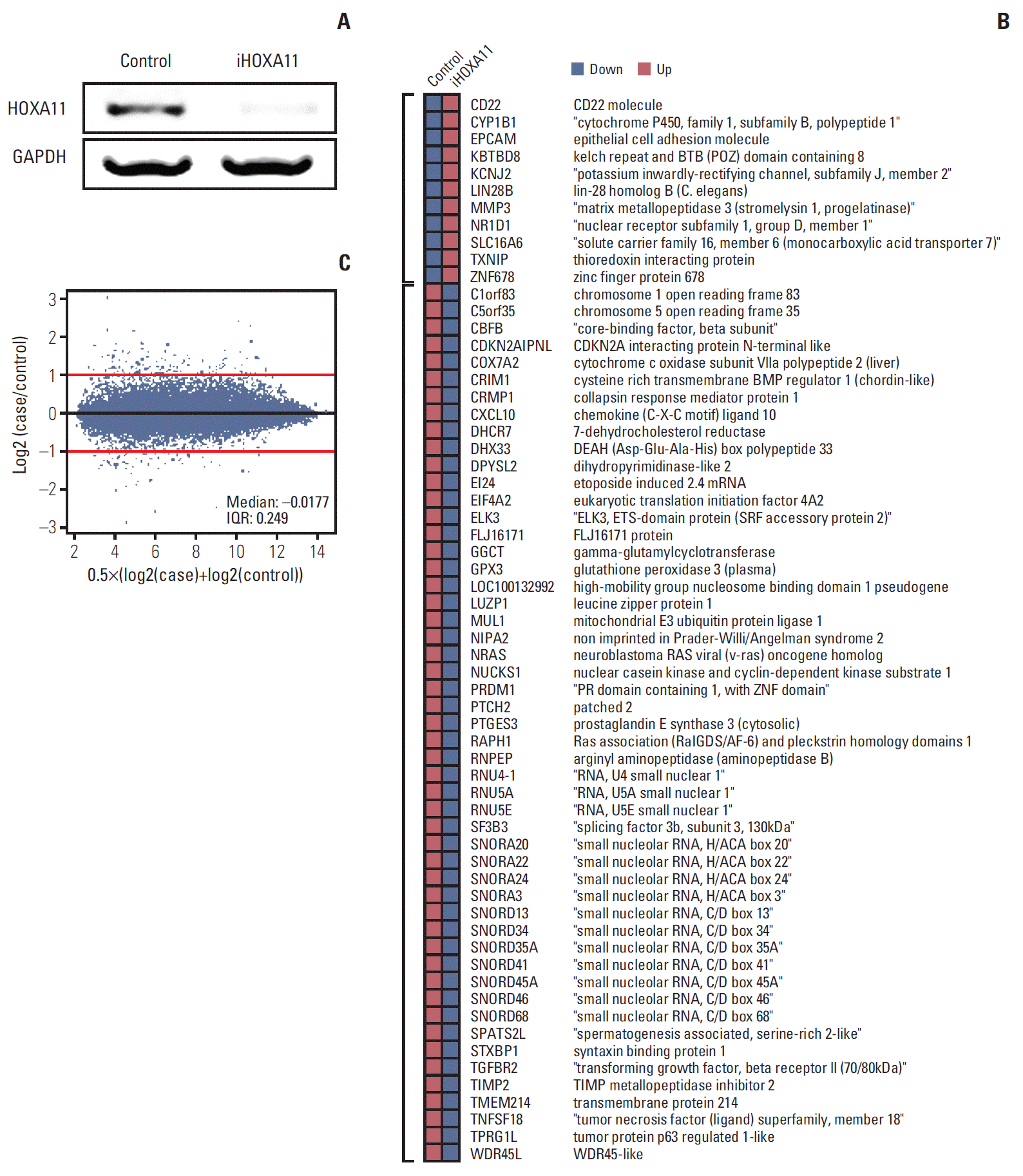
To identify the genes under the regulation of HOXA11, the microarray expression profiling data of the control LN18 cells was compared with that of the HOXA11-silenced cells with HOXA11 siRNA. LN18 was chosen because it showed the most significant decrease in HOXA11 expression after siRNA transduction among the three cell lines tested. After normalization of the values and DEG analysis, 11 up-regulated and 51 down-regulated genes that exhibited more than two-fold changes after HOXA11 suppression were identified (Fig. 4, Supplementary dataset).
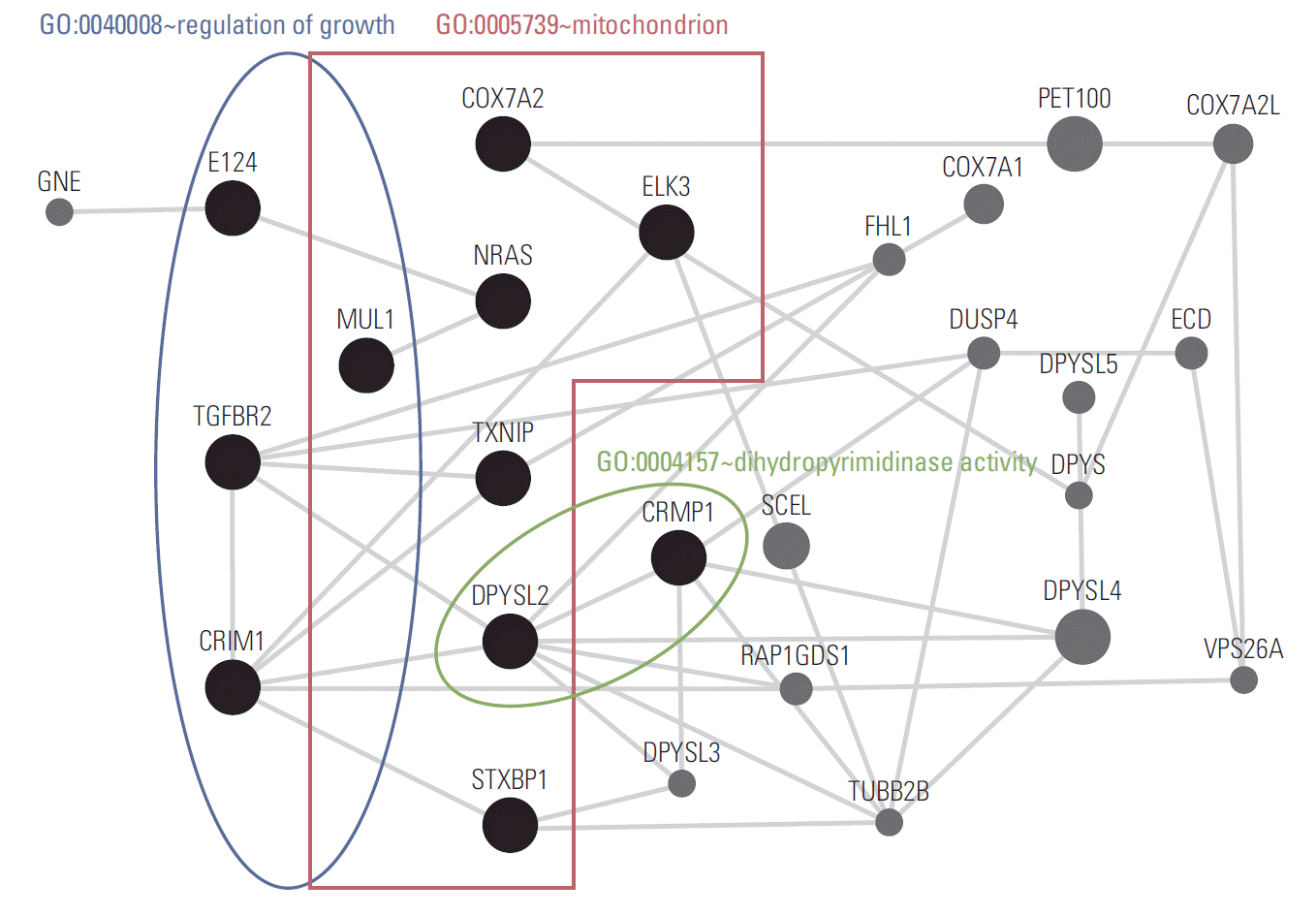
The functional annotation tools within DAVID Bioinformatics Resources (http://david.abcc.ncifcrf.gov/home.jsp) was then used for gene annotation enrichment analysis for the identified DEGs [19,21]. Three gene ontology categories (GO:0040008 regulation of growth, GO:0005739 mitochondrion, and GO:0004157 dihydropyrimidinase activity) were enriched significantly (EASE score < 0.05) in 11 genes (EI24, CRIM1, MUL1, TGFBR2, TXNIP, STXBP1, ELK3, COX7A2, NRAS, DPYSL2, and CRMP1) among the DEG set (Table 2). For deeper insight into the HOXA11 regulatory mechanism, network analysis was performed using GeneMANIA software (ver. 3.1.2.8) [20]. The network was constructed with the coexpression relationships, which revealed transforming growth factor, beta receptor 2 (TGFBR2), cysteine rich transmembrane BMP regulator 1 (CRIM1), thioredoxin interacting protein (TXNIP), dihydropyrimidinase-like 2 (DPYSL2), and collapsin response mediator protein 1 (CRMP1) genes to be the key hub regulators associated with HOXA11 suppression (Fig. 5).
Discussion
HOX genes, which are a cluster of master regulators of embryogenesis, are expressed temporarily during the developmental phase in vertebrates, but they should be silenced in the adult central nervous system [2,21,22]. Accumulating evidence of the aberrant expression of HOX genes in cancers suggests that these genes have diverse roles in oncogenesis [2,3,21-26]. In addition, a relationship between the HOX genes and treatment resistance or prognosis in cancer has frequently been proposed [9,11,12,27,28]. Several studies have revealed the tumor suppressor roles of HOX genes in many cancers [1]. Moreover, there is evidence suggesting that the restored expression of tumor suppressor HOX genes can attenuate the cancer progression in vitro and in vivo [29-31]. Experimental evidence of an association between HOXA10 and TMZ resistance in GBM has been presented [12]. Subsequent reports into the oncogenic role of HOXA10 in various cancers have been published [32-40]. On the other hand, based on the previous experimental data it is suspected that HOXA11 acts as a tumor suppressor in opposition to HOXA10, which prompted a further study of the function of HOXA11 in GBM.
The present study proposes a tumor suppressor function of HOXA11 in GBM based on the results from both in vitro experiments and human samples. The role HOXA11 in diverse cancers has been reported [41-47]. In gastric cancer, the epigenetic down-regulation of HOXA11 has been related to carcinogenesis, proliferation, migration, and invasion [41,42]. Similarly, in lung and ovarian cancers, the down-regulation of HOXA11 was shown to be a poor prognostic factor [43,44]. In GBM, the epigenetic down-regulation rate of HOXA11 was reported to be 51%-75%, and HOXA11 was one of the most frequently methylated genes in GBM [45-47]. The methylation of HOXA11 was associated with older patient ages and poor survival in GBM [47]. In addition to the evidence of the epigenetic characteristics of HOXA11 as a prognostic marker, the present study provides direct evidence of the prognostic value of HOXA11 expression in GBM samples (Figs. 1 and 2). Based on previous reports and the results in the present study, it is obvious that HOXA11 is a tumor suppressor in GBM and other cancers.
Treatment resistance induced by HOXA11 down-regulation, as detected by in vitro experiments in this study, is a mechanism contributing to a poor prognosis. In addition, this study detected candidate mediators (TGFBR2, CRIM1, TXNIP, DPYSL2, and CRMP1) that may impart treatment resistance after HOXA11 suppression. HOXA11 was reported to be a tumor suppressor gene that can sensitize a chemotherapeutic agent in ovarian cancer [48,49]. On the other hand, there is little direct evidence of treatment resistance induced by HOXA11 suppression in other cancer types. Nevertheless, these results provide a promising basis for the development of HOXA11 applications that target chemo- or radio-sensitizers in GBM. Among the five candidate mediators of HOXA11 suppression-induced oncologic effect, none have been reported to be associated with a HOXA11 regulatory mechanism. On the other hand, CRMP1 is notable because it is an invasion-suppressor gene in cancer cells [50]. Recently, it was suggested that decreased CRMP1 expression in GBMs harboring EGFRvIII positivity is responsible for promoting invasion [51]. Moreover, those authors proposed the counter-activation of Rac-1 after CRMP1 suppression as a mechanistic hypothesis for the invasive phenotype [51]. Other evidence regarding the role of HOXA11 in oncogenesis includes a report on the regulation of matrix metalloproteinase 2 expression, which can affect cancer cell migration and invasion [52]. Overall, this study postulates an oncogenic pathway involving EGFRvIII, HOXA11, CRMP1, and Rac-1 in GBM, which will require further investigation.
 XML Download
XML Download