

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Diabetes is an irreversible disease, identification of risk factors and disease prevention are therefore important for its management. Previous research has revealed risk factors for type 2 diabetes mellitus (T2DM) including body mass index (BMI), waist circumference, blood pressure, family history, and genetic background [12].
Many investigators have assumed that identification of genetic factors will improve the prediction of T2DM, so many genetic studies have been performed [3]. With the advent of high-throughput genotyping technologies, genome-wide association studies (GWASs) have become prevalent [4]. Several hundreds of thousands of tag single nucleotide polymorphisms (SNPs), which represent genotype variation of neighboring SNPs, are selected and probes for the tag SNPs are used in SNP microarray chips. Usually, more than 10,000 samples are used and imputed with a reference panel from a population sequencing project such as the 1000 Genome Project [5]. These GWASs have been actively applied to the study of T2DM, and meta-analysis of the individual GWASs identifies genome-wide loci associated significantly with the development of T2DM [36]. For example, Unoki et al. [7] reported that rs2283228, which is located in potassium voltage-gated channel subfamily Q member 1 (KCNQ1), shows genome-wide significance and the result was replicated in a different population [7]. In a meta-analysis of GWASs of European populations, 12 novel loci that are enriched in genes involved in cell cycle regulation have been identified [8]. A trans-ethnic meta-analysis of several consortia for a GWAS of diabetes revealed a directional consistency of the risk alleles across different ethnic groups and seven newly identified susceptible loci for T2DM [9]. While these studies used SNP microarray data, a sequencing-based GWAS and combination with the chip-based data were applied recently [10]. The results showed that variants associated with T2DM are located within the region that identified with previous GWASs.
Although the exome covers only about 1% of the entire human genome, it is easier to find functional implications for the significant loci because there is a lot of information about gene function. In genetic studies of T2DM and related traits, the exome-wide association studies have been performed with an exome array or exome sequencing to identify susceptible loci [1011121314].
In the present research, using exome microarray data from 14,026 Koreans, we identified coding variants having susceptibility for T2DM. We also applied the genotype information of the variants into a model to predict T2DM and tested the performance of the model in the entire population and subgroups stratified by BMI.
METHODS
Study population
We used epidemiological data from the Ansan/Ansung (ASAS), Cardio-Vascular disease Association Study (CAVAS) and Health Examinee (HEXA) cohort, which are part of the Korean Genome and Epidemiology (KoGES) project [15]. The present study was approved by the Institutional Review Board of Korea Centers for Disease Control & Prevention (2017-03-10-1C-A) and written consent was obtained from all participants. The participants in the ASAS cohort underwent an oral glucose tolerance test (OGTT) using a solution of 75 g glucose. Using the OGTT results, we defined T2DM patients according to their past history of diabetes or American Diabetes Association criteria [16], which include (1) fasting glucose ≥126 mg/dL, (2) 2-hour glucose ≥200 mg/dL, and (3) glycosylated hemoglobin ≥6.5%. The OGTT had not been used in participants from the CAVAS and HEXA cohorts, and so the history and fasting glucose level were used to determine diabetes status in these cohorts.
Exome chip genotyping and quality control
We used the exome chip data that had been generated in a previous study [17]. Detailed genotyping and quality control processes were described previously. In brief, the Illumina HumanExome Chip v1.1 (Illumina Inc., San Diego, CA, USA), which includes probes for 242,901 variants, was used for genotyping. Illumina GenomeStudio version 2011.1 software clustered the probe signals to determine the genotype. After genotype calling, variant filtering was performed based on the following criteria: (1) completely missing in all participants, (2) monomorphic SNP, (3) Hardy-Weinberg equilibrium P<1.0× 10−6, and (4) genotype call rate of <95%. Samples with cryptic relatedness and sex discrepancy were also excluded. After filtering, we imputed a missing genotype using the phasing function of the Beagle software [18].
Genome-wide association analysis
We used PLINK 1.9 for GWAS of the exome chip and meta-analysis [19]. The logistic function was applied with covariates of sex, age, and BMI. We used the adjust option to estimate λ for genomic control of GWAS statistics. For meta-analysis, we used the meta function of PLINK, and the result was filtered with Cochrane Q statistics and the I2 index. Bonferroni's multiple testing correction was applied to select exome-wide significant loci. To search published GWAS results, we used the Korean Reference Genome Database (KRGDB) [20].
Prediction analysis and comparison of performance
Using follow-up information from the ASAS cohort data, we predicted T2DM using clinical and genetic variables. The glm function of the R statistical software package was used to develop the predictive model using follow-up information for T2DM status [21]. To compare different models, we used the area under the curve (AUC) and continuous net reclassification improvement (cNRI). To estimate the AUC, we used the partial receiver operating curve component of the R package [22]. The cNRI was estimated and tested statistically using the reclassification function of the PredictABEL R package [23].
RESULTS
Study population
The age of the study population ranged from 39 to 89 years, and mean±standard deviation (SD) was 54.22±9.23 years (Table 1). The number of female participants (n=7,588) was higher than that of male participants (n=6,438). The mean±SD of the BMI was 24.37±3.09, and the BMI of 5,561 participants (40%) was over the threshold for being overweight (≥25). Among the 14,026 participants to whom the exome array was applied, there were 1,992 (14%) patients with T2DM. The three covariates (sex, age, and BMI) that were used in the GWAS were significantly different between the group with T2DM and the control group (P<0.05) (Supplementary Table 1).
Result of the exome-wide association study
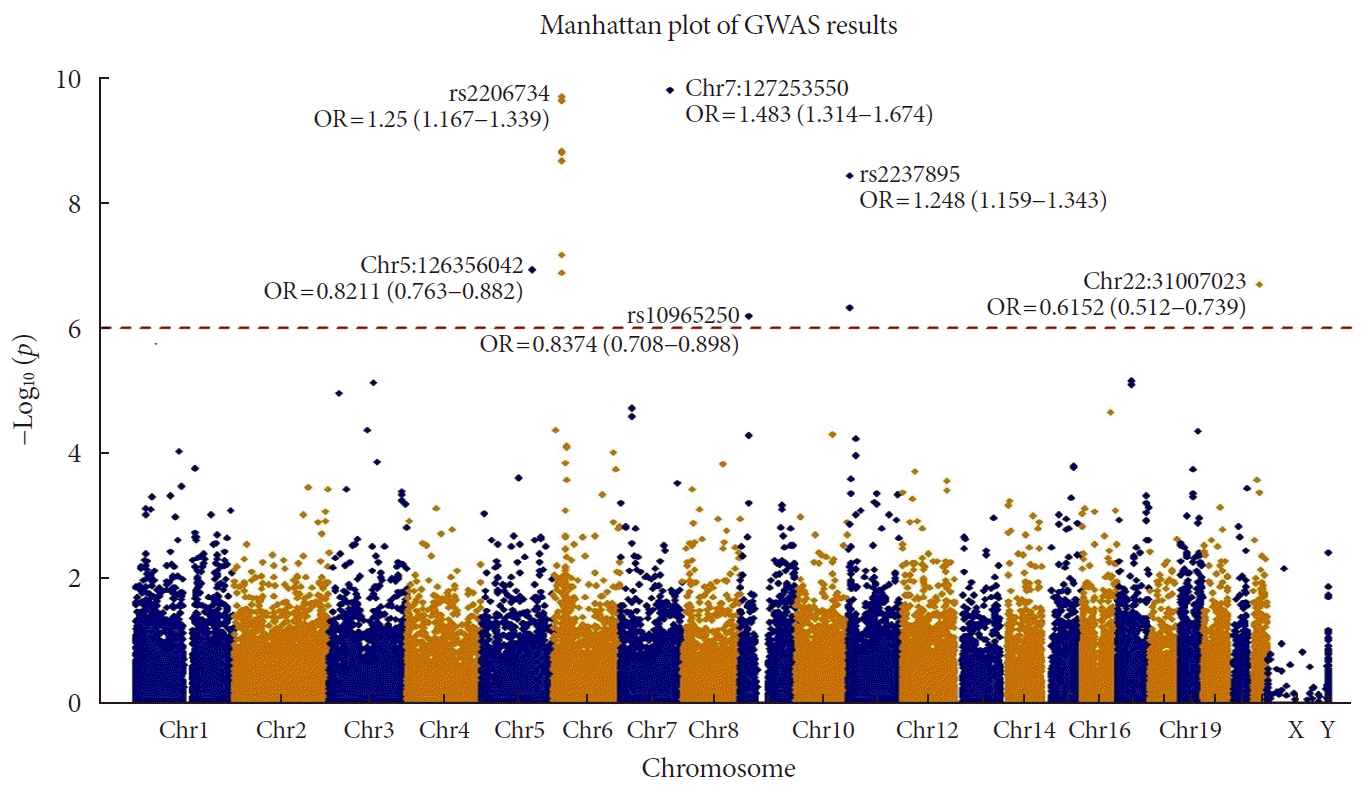
From the AS, HEXA, and CAVAS cohorts, exome chip data of 7,461, 3,456, and 4,566 participants were used in the analysis. After filtering, 50,840 variants were tested for their genetic susceptibility to T2DM. The genomic inflation factor was 1.00, which indicated that there was no inflation of the GWAS result. In the quantile-quantile plot, most of the variants showed null distribution, and a few loci that had a significant association with T2DM showed a marked deviation from the diagonal line (Supplementary Fig. 1).
When we applied Bonferroni's multiple testing correction to the results of GWAS using logistic regression with the additive genetic model, 15 variants showed exome-wide significance with cut-off P=1.9.83×10−7 (0.05/50,840). We used the PLINK clump function to filter the variants based on linkage disequilibrium (LD) and distance between variants. We applied default parameters with LD=0.2 and distance=250K. After the clumping, we obtained six variants (Table 1 and Fig. 1). Among them, rs2233580 showed the highest significance (P=1.60×10−10) with an odds ratio (OR) of 1.48 (95% confidence interval [CI], 1.31 to 1.67), which is located on paired box gene 4 (PAX4). The rs2206734, which is located on cyclin-dependent kinase like 1 (CDKL1), showed the second highest significance (P=1.60×10−10). The rs2237895 of KCNQ1 gene was identified as susceptible loci only in the GWAS of an East Asian population, and it was replicated in our result. The rs11960799 has not been reported as a susceptible locus for T2DM, although it has shown nominal significances for triglyceride level, rheumatoid arthritis, and birth weight previously [24]. The OR of the variant was less than 1 (=0.82), which indicated a protective effect for T2DM. The rs75680863 also showed a protective effect (OR, 0.61; 95% CI, 0.51 to 0.74) for T2DM. This variant was a novel locus that had never been reported for its association with the development of T2DM. rs2237892 also showed a protective effect against the development of T2DM (OR, 0.83; 95% CI, 0.77 to 0.89). It is also located in KCNQ1, and previous studies reported that this variant had genome-wide significance in its association with T2DM [725]. When nominal significance (P<0.05) is considered, there are a lot of significant associations between T2DM and the other traits and the rs2237892 variant (Supplementary Fig. 2). Interestingly, the ORs of the previous studies of GWAS for T2DM are >1, which indicates increasing risk of disease. rs10965250, having the least significant P value, appears to have a protective effect against T2DM. The variant has been reported to have a genome-wide association with T2DM in a European population [8].
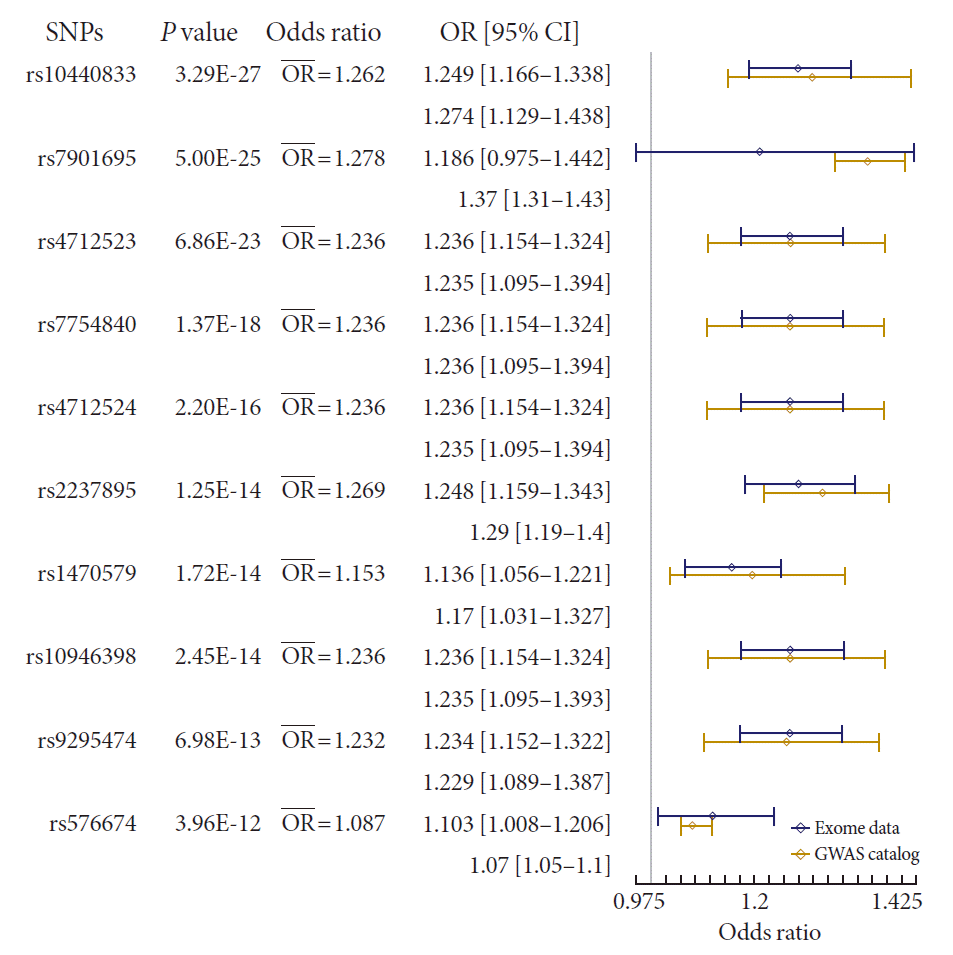
We also performed a meta-analysis using our results and summary statistics from the GWAS catalog database. In the GWAS catalog, we extracted 1,222 variants having genome-wide significance (P<0.5×10−8) in susceptibility for T2DM. Among them, 124 variants were identified in our exome array data. We used the variants in a meta-analysis, and the summary statistics of the variants from our results and the GWAS catalog were applied to the meta-analysis. The P values from a fixed effects model were used to test significance. We used a P threshold of 9.83×10−7, which was used to identify significant exome-wide results in our data. To exclude a heterogeneous effect of the variants under the fixed effects assumption, we selected results with a threshold of I2 <50% and P of Cochrane Q statistics >0.05. After filtering, 23 variants were found to be significant (Fig. 2 and Supplementary Table 2). rs10440833, which lies in CDKL1, showed the most significant result (P=3.27×10−27). Interestingly, of the exome-wide significant loci in our data, only rs2237895 showed significance in the meta-analysis (P=1.25×10−14) (Supplementary Table 2).
Prediction of future diabetes using clinical and genetic markers
Using significant loci in the exome-wide association analysis, we performed prediction analysis. In the prediction analysis, we used clinical variables including fasting glucose, BMI, high-density lipoprotein, sex, parental history of diabetes, triglyceride, systolic pressure, diastolic pressure, and treatment status of hypertension. These have been frequently used to develop models to predict T2DM [26]. The incident diabetes was identified using 10-year follow-up information of the ASAS cohort. The number of baseline participants who did not have T2DM was 7,889 and 6,317 participants had exome chip data. In total, 1,054 participants having exome chip data developed T2DM during follow-up.
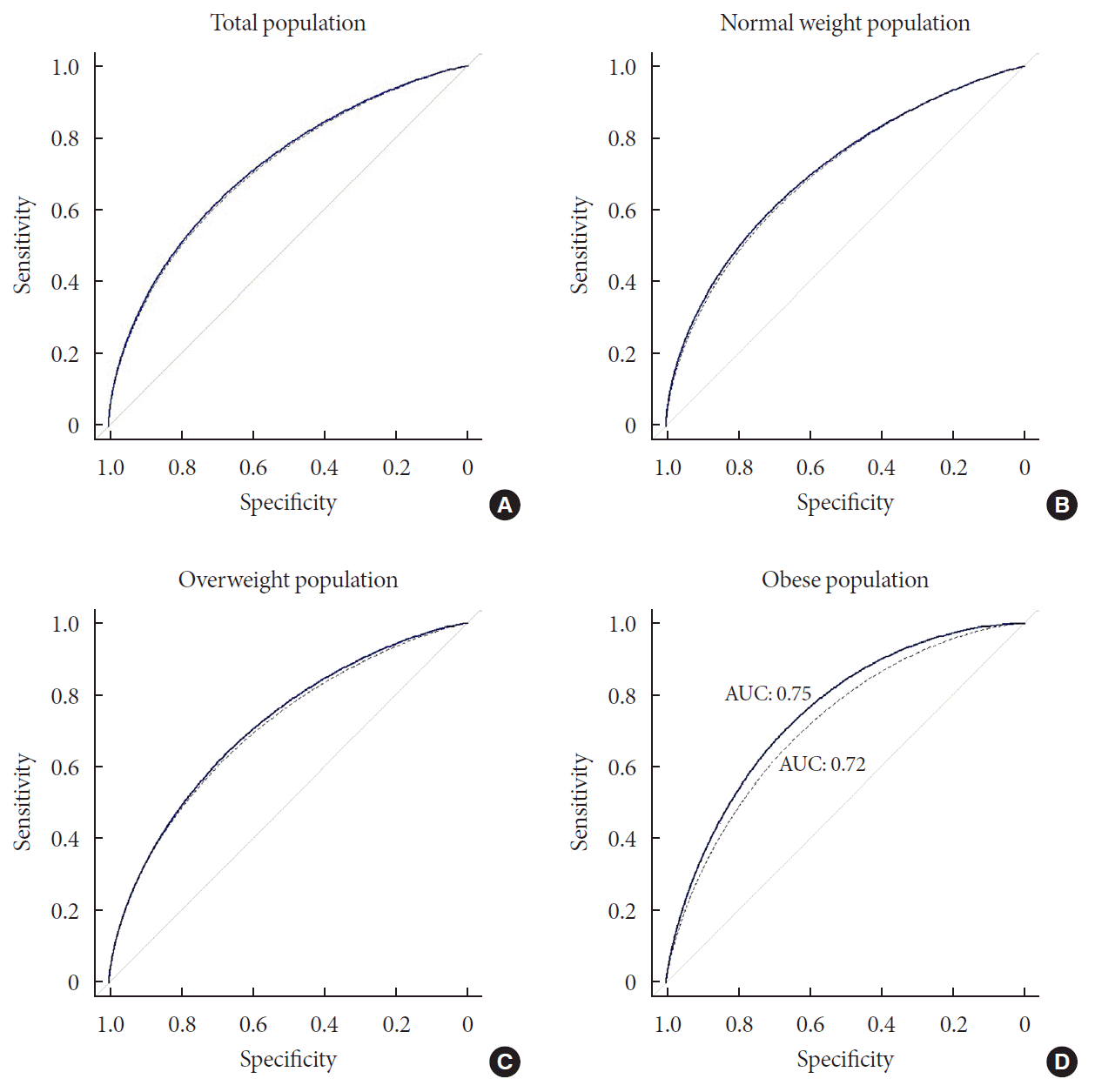
To measure the predictive performance, we estimated the AUC and the net reclassification index (NRI) was used to compare the prediction models with or without genetic information (Fig. 3). When the prediction model was built using the clinical variables, the AUC was 0.71 (95% CI, 0.70 to 0.73). The AUC increased to 0.72 (95% CI, 0.70 to 0.74) with the genotype information of the 7 loci. The difference in the AUCs was significant (P=1.04×10−3) and the NRI (0.11) was also significant (P=9.80×10−4).
We then compared the prediction performances of the clinical variables and genetic markers between BMI subgroups. We divided the total population into three BMI subgroups: normal (BMI <25, n=3,727), overweight (25≤ BMI <30, n=2,314), and obese (BMI ≥30, n=267) individuals. We applied logistic regression to each subgroup and compared the prediction results with the analysis of the entire population. We found that there was a substantial difference in the prediction performance for future T2DM between the BMI subgroups (Fig. 3). In the normal group, AUC of the clinical model and combined model (clinical+genetic information) was 0.70 (95% CI, 0.68 to 0.73) and 0.71 (95% CI, 0.69 to 0.74), respectively. The difference in AUC was significant (P=0.03). The NRI was 0.14 (95% CI, 0.05 to 0.23), which was also significant (P=3.58×10−3). In the overweight group, there was no significant difference in AUC between clinical model and combined model (0.72 and 0.73, respectively), although the NRI was significant (NRI=0.12; 95% CI, 0.02 to 0.22; P=0.02). The difference in AUC between the clinical model and combined model was highest for the obese group (Fig. 3). Although not significant (P=0.16), the AUC of the combined model increased to 0.75 (95% CI, 0.69 to 0.81), compared with that of the clinical model in the obese group, which was 0.72 (95% CI, 0.65 to 0.79). Moreover, the NRI was 0.52 (P=8.00×10−5); that is, 4.3-fold that of the overweight group, which had the lowest NRI value. To exclude the possibility that the greater AUC in the combined model in the obese group resulted from a small sample size, we randomly selected the same number of participants as those in the obese group (n=267) and estimated the AUC. We repeated this process 1,000 times. No AUC from random selection of the participants showed a greater AUC than that of the obese group (Supplementary Fig. 3).
DISCUSSION
Here we identified the exome-wide loci having susceptibility for T2DM in a Korean population. When we applied the loci to the predictive model for T2DM, we found that while performance of the model was marginal in the total population, the performance was more obvious in the obese group.
rs2233580 had the highest OR (1.45; 95% CI, 1.31 to 1.67; P=1.60×10−10). This variant had been reported in a recent exome-wide association study using exome sequencing data, and the current data were used to replicate the analysis [27]. The variant is located in PAX4, which is a transcription factor involved in pancreatic islet development [28]. In previous studies, variants of PAX4 were found to be associated with T2DM [162930]. In particular, recent genome-wide or exome-wide association studies with East Asian populations revealed PAX4 variants are associated with T2DM [192132]. In the present study and in an exome chip analysis of T2DM in a Chinese population, rs2233580 had the highest OR for significant variants in each study (1.45 and 1.38 in Korean and Chinese populations, respectively) [12]. PAX4 has a potential role in the management of T2DM, especially by preserving or replenishing β-cells [28]. It was notable that no significant results for rs2233580 have been reported in GWAS of non-Asian populations. rs2237892 and rs2237895, which are located in KCNQ1, are also well-known for their genome-wide significant association with the development of T2DM (Supplementary Fig. 4). Interestingly, the significance of rs2237895 has been reported only for an East Asian population, while rs2237892 and other variants of KCNQ1, such as rs2237896 and rs2237897, were significant in other ethnic groups (Supplementary Table 3). The rs2206734 lies in cyclin-dependent kinase 5 regulatory subunit associated protein 1 like 1 (CDKAL1), and the SNP has been reported in many GWASs of T2DM or related traits such as fasting glucose level and BMI. When we searched the variant on the KRGDB, we found many associations with metabolic traits including T2DM (Supplementary Fig. 5). rs10965250 is located in the intergenic region between AL157937.1 and CDKN2B-AS1 and has been reported to be associated with T2DM and fasting glucose, 2-hour glucose level, waist-hip ratio, and birth weight in various ethnic groups (Supplementary Fig. 6). The two variants (rs2237892 and rs10965250) had OR <1, indicating the protective effect of the variants in the development of T2DM. However, the previously reported ORs were >1, indicating the ethnic-specific effect of the variant. This finding should be confirmed in future studies.
rs11960799 within membrane associated ring-CH-type finger 3 (MARCH3) located at 126,356,042 of chromosome 3 had never been reported for its association with T2DM. In previous studies, rs11960799 showed a significant association with triglyceride level, rheumatoid arthritis, and birth weight. However, these variants were found to have nominal significance (P<0.05), and did not reach genome-wide significance. Considering a previous report that showed the significant transcript change of MARCH3 in human islet cells [31], it seems possible that rs11960799 is at least a variant associated with the development of T2DM. rs75680863 is another variant newly discovered in the present study. No previous GWASs have ever reported the significant association of this variant and T2DM. The OR of rs75680863 was 0.63, which is equivalent to an OR of 1.58 if the direction of the coefficient of the variant is positive, and the size of the OR is highest among those of the significant variants. rs75680863 lies within transcobalamin 2 (TCN2), the gene for transcobalamin 2, which is a member of the vitamin B12 binding protein family. Because vitamin B12 deficiency is closely related to T2DM [32], there is a possible pathway involving vitamin B12 metabolism and the development of T2DM.
In the present meta-analysis, we identified 32 loci showing significance, and the levels were sufficiently high to reach genome-wide significance (P=1.5×10−8). In a fixed effects model, more variants (n=64) among the 124 that were used in the meta-analysis showed significance. However, after filtering based on Cochrane Q statistics and the I2 heterogeneity index, the number decreased to 32. Moreover, only a single significant variant from our results was included in the significant result from the meta-analysis. These might imply substantial heterogeneity in the genetic effect for development of T2DM in Koreans.
In this study, we performed prediction analysis using 7 significant loci. While many studies used genetic risk score (GRS) by summing the risk scores of individual loci, we used the 7 loci directly because the number of loci was not so high. When we applied the 7 significant loci to the prediction analysis, we obtained a significant, but marginal, increase in the AUC. This result was consistent with those of previous studies [33]. These results were consistent regardless of the number of genetic markers that were used in the prediction models. For example, Vassy et al. [34] used different numbers of common genetic variants for predicting incident T2DM. Even when genetic information was integrated into the prediction model, the performance showed only marginal improvement. Talmud et al. [35] reported similar results with weighted scoring of 65 common genetic variants. They showed a significant but marginal change of receiver operating curve (ROC) (from 0.75 to 0.76, P=0.0003) with integrating a GRS into their prediction model. Lall et al. [36] applied a greater number of genetic variants into a prediction model. They used 1,000 SNPs for developing a prediction model using a doubly weighted GRS method. In the results, the prediction model with the GRS showed a slight increase of AUC (from 0.77 to 0.79), but highly significant P value (2.01×10−11) of the difference of AUC between the model with or without GRS [36]. The NRI was mildly but significantly increased in our results; this compares to previous studies that also reported a mild increase of continuous NRI in a model with genotype information [343536]. This seems to indicate that one clinical implication of a genetic prediction model of incident T2DM lies with the reclassification of the at-risk population, not with lifting overall prediction performance.
When we divided the total population into three groups (normal, overweight, and obese), the results of prediction analysis for incident T2DM differed in the obese group compared with the other groups. The AUC and NRI changed slightly in the normal and overweight groups, but the performance measures showed greater changes in the obese group. Although the AUC of the prediction model with genetic information showed nonsignificant change, the magnitude of the change of the AUC was greater than for the other subgroups (Fig. 2). Moreover, the NRI was highly significant and far greater than the NRI of the other subgroups. The change of prediction performance in different subgroups of a population have been reported in many GWASs. In particular, such results were found in BMI subgroups. Previous studies reported similar findings in terms of different association and prediction performance in BMI subgroups. Morris et al. [38] found that rs4506565 near transcription factor 7-like 2 (TCF7L2) had different associations in a nonobese group (BMI ≥30; OR, 1.54) and an obese group (BMI >30; OR, 1.20). In another GWAS for T2DM using BMI subgroups [39], rs8090011 in LAMA1 had genome-wide significance in lean individuals (BMI <25; P=8.4×10−9) and not in obese individuals (BMI ≥30; P=0.04). Timpson et al. [40] also reported that rs8050136 of fat mass and obesity-associated protein (FTO) gene was detectable in the obese group (BMI ≥30.2; P=1.3×10−13), while the significance disappeared in the nonobese group (BMI <30.2; P=0.19). Besides significance, prediction performance differs according to the BMI subgroups. In the prediction analysis of T2DM with GRS of 65 common variants, samples of the lowest tertile (BMI <24.5) had an NRI of 27.6%, while those of the upper tertile (BMI >27.5) had an NRI of 2.6% [35]. Previous studies showed that the prediction model with GRS outperformed in the lean body group than in the obese group, but our result was different. The difference of AUC and magnitude of reclassification was greater in the obese group. While GWASs have been identifying susceptible loci for diabetes, previous studies have shown that integration of genetic information or GRS from GWAS results increased the performance of diabetes prediction marginally [33]. However, we found that the prediction performance increased in obese group more than in whole population. In the obese group, the clinic-genetic prediction model increased AUC marginally, but cNRI significantly (P=8.00×10−5). Moreover, permutation test revealed that the AUC of prediction model of obese group is significant (Supplementary method). These results indicate that the clinic-genetic model of obese group increased overall prediction performance marginally, but re-assign risk probability to patients more accurately. Clinically, it would be helpful when patients need to be matched to a preventive protocol because risk assessment is more accurate. Considering obese group had limited number of population in general, it might be cost-effective to apply such model to the specific subgroup of population.
These seemed to indicate that the genetic information may be more valuable predictor of diabetes in a specific subpopulation. The identification of such groups should be investigated in further research. In addition, each loci showed the same tendency of high performance of prediction in obese group (Supplementary Table 4). This might result from our use of exonic variants in the prediction model. We thought that the change of prediction accuracy between BMI subgroups results from epigenetic change of the loci. It is reported that the KCNQ1 gene of blood cells is hyper-methylated in obese population [37]. If so, it is also possible that KCNQ1 gene of diabetes-related cells can be hyper-methylated, which might result in change of KCNQ1 gene expression, and prediction accuracy of the loci eventually. Considering that KCNQ1 contains two loci of the significant results in this analysis, it is possible that higher AUC is associated with methylation change, and the other loci might also have the methylation changes nearby the loci, which affect the prediction accuracy in obese group.
In summary, we found significant exonic variants for T2DM, which were previously identified or newly detected. Genetic prediction using the loci showed a mild but significant increase of AUC and NRI. We found that the variants had higher performance in the obese group than in the normal and overweight groups. We believe that our results provide clues to reveal the genetic architecture of T2DM in the Korean population.
 XML Download
XML Download