

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The coronavirus disease 2019 (COVID-19) is caused by the novel coronavirus severe acute respiratory syndrome coronavirus-2 (SARS-CoV-2) and has spread globally causing international concerns.123 As of August 23, 2020, there were 23,057,288 confirmed cases of COVID-19 in 216 countries, with 800,906 confirmed deaths.4 Most patients are asymptomatic or recover after mounting a self-limiting antiviral response with the development of neutralizing anti-viral antibodies and cell-mediated immunity.5 However, around 10% of all cases become serious, with dyspnoea, lymphopenia, and extensive chest X-ray abnormalities and half of these become critically ill, with respiratory and multi-organ failure.678 There appears to be a relationship between the clinical and immunological features of COVID-19, as the disease severity correlates with certain immunological markers.79 Recent studies have shown that severe and critically ill patients exhibit ‘cytokine storm’, which is related to the production of excessive cytokines, dysregulated immune cell function, and massive systemic inflammation.1510 Understanding the causes of altered immune features of COVID-19 would enable the refinement of preventive vaccine targets and accelerate therapeutic development. Despite this, the molecular mechanisms underlying exaggerated inflammatory phenotypes during COVID-19 are largely unknown.
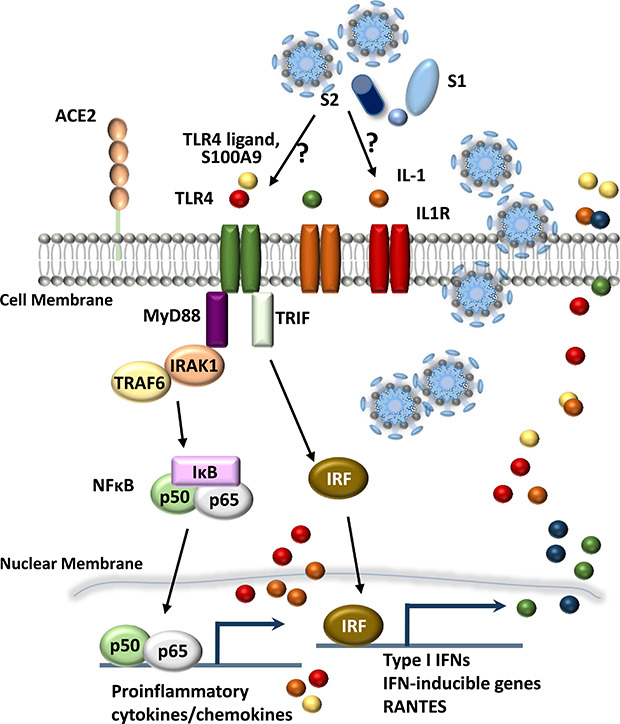
SARS-CoV-2 belongs to subfamily Coronavirinae in the family Coronaviridae.11 The spike (S) glycoprotein, which is immunogenic to produce antibodies and crucial for the entry into host cells, harbors a furin cleavage site between the S1/S2 subunits.12 Recent efforts for designing epitope-based peptide vaccine based on an immune-informatics approach showed that a multivalent subunit vaccine targeting S2 subunit of SARS-CoV-2 S glycoprotein might have potential to activate innate and adaptive immune responses.13 In addition, the receptor binding domain (RBD) of the S protein of SARS-CoV-2 appears to be potentially useful in the serological diagnostic assays for COVID-19 patients.14 Furthermore, the immune responses to S-RBD binding antibodies exhibited a correlation with neutralizing capacity, suggesting a potential COVID-19 immunity.15 Thus it is challenging whether each recombinant protein antigen of SARS-CoV-2 is able to induce innate immune responses in human monocytes and/or peripheral blood mononuclear cells (PBMCs).
In this study, we examined the immune-related transcriptome profiles in a total of 48 subjects including 28 COVID-19 patients, constituted with 20 mild/moderate (MILD) and 8 severe/critical (SEVERE) cases, and 20 healthy controls (HC). We found that toll-like receptor (TLR) 4-mediated inflammatory signaling molecules, which mimic pathogenesis of bacterial sepsis, are upregulated in PBMCs from COVID-19 patients. Although there was no significant immune biomarker between mild and severe groups, S100A9, an alarmin and TLR4 ligand, was the most highly increased inflammatory mediators in SEVERE patients, when compared to HC. Notably, it inversely correlated with the serum albumin levels. We also showed that recombinant S2 and nucleocapsid (NC) proteins of SARS-CoV-2 significantly increased pro-inflammatory cytokines/chemokines and S100A9 in human primary PBMCs. Finally, we showed that S2 protein in presence of recombinant S100A9 significantly amplified the IL1B mRNA expression in PBMCs, as compared to those stimulated with either S2 protein or S100A9.
METHODS
Study population
COVID-19 patients were confirmed by real-time quantitative polymerase chain reaction (RT-qPCR) for SARS-CoV-2 in nasopharyngeal and oropharyngeal swab, with or without sputum. Patients were categorized into two groups; MILD vs. SEVERE cases. In severity assessment, the World Health Organization's COVID-19 disease severity definition was used.16 Twenty-eight COVID-19 patients (8 SEVERE vs. 20 MILD) admitted to Chungnam National University Hospital, and age/sex-matched 20 HC, giving specific informed consent were included in the study. We excluded patients with age under 19. In the SEVERE group, two patients were transferred from a long term care facility in which had a COVID-19 outbreak. They had been hospitalized with well-controlled schizophrenia. Another patient was referred to our hospital in a state of endotracheal intubation. The patients' characteristics, clinical symptoms and laboratory test results are summarized in Table 1. All clinical and laboratory parameters were those at the time of sampling. The sampling point (median 5–6 days after illness onset) was determined by previous reports about COVID-19 patients,317 which was relatively early in the clinical courses. In asymptomatic patients, a screened date for COVID-19 because of strong epidemiologic link was used for illness onset.
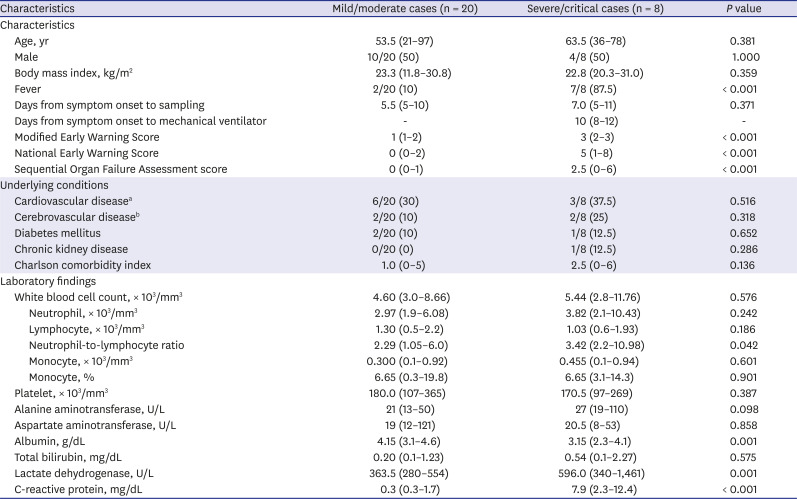
Table 1
Characteristics and laboratory findings of patients with COVID-19
Data are presented as medians (ranges) or numbers (%). For asymptomatic patients in the mild/moderate group, a time of diagnosis with COVID-19 was used as illness onset.
COVID-19 = coronavirus disease 2019.
a Including hypertension; bIncluding dementia and schizophrenia.
![]()
Nanostring nCounter assay
Nanostring nCounter Human Immunology gene expression assays and Human miRNA expression assays were performed at PhileKorea Technology (Daejeon, Korea), using the NanoString nCounter GX Human Immunology Kit V2 (NanoString Technologies, Inc., Seattle, WA, USA). Normalization of gene expression levels was performed by scaling with the geometric mean of the built-in control gene probes for each sample. Differentially expressed immune genes (DEiGs) among HC, SEVERE, and MILD patients satisfied false discovery rate (FDR) < 0.05 which was analyzed and corrected by wilcox.test and p.adjust functions, respectively, implemented in stat package of R (v. 3.6.2; R Foundation, Vienna, Austria).
Cell culture and SARS-CoV-2 recombinant protein stimulation
Human PBMCs from healthy volunteers were isolated from heparinized venous blood using Ficoll-Hypaque (Lymphoprep; Alere technologies, Oslo, Norway) as described previously.18 For monocyte-derived macrophages (MDMs) differentiation, adherent monocytes were incubated in Roswell Park Memorial Institute 1640 medium (Lonza, Basel, Switzerland) containing 5% pooled human serum (Sigma-Aldrich, St. Louis, MO, USA), 1% L-glutamine, for 1 hour at 37°C, after which the nonadherent cells were removed. Human MDMs were prepared by culturing peripheral blood monocytes for 4 days in the presence of 4 ng/mL human macrophage colony-stimulating factor (R&D Systems, Minneapolis, MN, USA). SARS-CoV-2 (2019-nCoV) NC-His recombinant protein (cat. No. 40588-V08B), Spike S1-His recombinant protein (cat. No. 40591-V08H), Spike S2 extracellular domain (ECD)-His recombinant protein (cat. No. 40590-V08B), and Spike RBD-His recombinant protein (cat. No. 40592-V08H) were purchased from Sino Biological, Beijing, China. Cells were stimulated with the proteins as indicated in figure legends.
RNA extraction and RT-qPCR
Total RNA from PBMCs or MDMs was extracted using QIAzol lysis reagent (Qiagen, Hilden, Germany) and miRNeasy Mini Kits (Qiagen) according to the manufacturer's instructions, followed by RNA quantitation. cDNA from total RNA was synthesized using the reverse transcription master premix (ELPIS Biotech, Daejeon, Korea). RT-qPCR was performed in Rotor-Gene Q 2plex system (Qiagen) using SYBR green master mix (Qiagen) and primers for indicated genes. Primers used in this study are listed in Supplementary Table 1. Data were analysed using 2ΔΔ threshold cycle method where GAPDH was used for normalization.
Immunoblot analysis and enzyme-linked immunosorbent assay (ELISA)
Cells were lysed using RIPA buffer (ELPIS Biotech) containing protease and phosphatase inhibitor cocktails (Roche Diagnostics, Mannheim, Germany) and equal amount of protein mixed with sodium dodecyl sulphate sample buffer were boiled for 5 minutes. Samples were subjected to sodium dodecyl sulphate-polyacrylamide gel electrophoresis and then transferred to polyvinylidene difluoride membrane. The membranes were blocked in 5% skim milk in Tris-buffered saline containing 0.1% Tween 20 (TBS-T) for 1 hour at room temperature, and then incubated overnight with following primary antibodies at 4°C: phospho-nuclear factor (NF)-κB p65 (Ser536) from Cell Signaling Technology (Danvers, MA, USA) and beta-actin from SantaCruz Biotechnology (Dallas, TX, USA). Membranes were washed using TBS-T and further incubated with appropriate secondary antibodies (Cell Signaling Technology) for 1 hour at room temperature. The immune-reactive proteins were detected using a chemiluminescence kit. ELISA to detect the levels of interleukin (IL)-6 in cell supernatant was performed according to the manufacturer's protocol (R&D Systems; cat. No. DY206-05).
Bioinformatics analysis
Spearman's correlation coefficients of gene expression levels of 579 immune genes were calculated with a cor.test function implemented in stat package of R. The KEGG pathway enrichment analysis was performed using DAVID (version 6.8; https://david.ncifcrf.gov) with a human reference gene set. We picked out significantly enriched pathways with FDR < 0.05.
To identify chemokine, IL, tumor necrosis factor (TNF), interferon (IFN) and those receptor gene families, we downloaded gene family annotations from HUGO Gene Nomenclature Committee (https://www.genenames.org).19
Statistical analysis
Statistical analyses were performed with Analyse-it, version 5.1 (Analyse-it Software, Ltd., Leeds, UK), SPSS Statistics for Windows, version 24.0 (SPSS Inc., Chicago, IL, USA), and GraphPad Prism, version 5.0 (GraphPad Software, San Diego, CA, USA). The data were processed by principal component analysis (PCA), Spearman's correlation, Student's t-test, Mann-Whitney U test, analysis of variance, and Kruskal-Wallis H test, as appropriate, and detailed in each figure and figure legends. Results are presented as medians (ranges) or means ± standard error of the mean or ± standard deviation (SD) as indicated in figure legends.
Ethics statement
This study was approved by the Institutional Research and Ethics Committee at Chungnam National University Hospital (Daejeon, Korea; CNUH 2019-04-046, CNUH 2020-07-082) and conducted in accordance with the Declaration of Helsinki.20 Informed consent was submitted by all subjects when they were enrolled.
RESULTS
Characterization of immune features of COVID-19 patients in terms of clinical severity
To investigate the immune signaling signature of COVID-19, a total of 48 Korean subjects (untreated COVID-19 patients with various clinical severities [n = 28] and HC [n = 20]) were enrolled in the study. Table 1 summarizes the characteristics and laboratory findings of 20 MILD (median age 53.5 [range 21–97] years) and 8 SEVERE (median age 63.5 [range 36–78] years) patients. In the SEVERE group, 7 of 8 (87.5%) patients had fever at the time of sampling vs. only 2 (10%) in the MILD group. The median time from symptom onset to mechanical ventilation was 10 (range 8–12) days. Underlying comorbidities were present in about half of the patients (hypertension, diabetes mellitus, dementia, schizophrenia, and chronic kidney disease) and did not differ between the groups. The median time from symptom onset to sampling was 5.5 (range 5–10) and 7 (range 5–11) days in the MILD and SEVERE groups, respectively. The scores for the degree of illness were higher in the SEVERE group at the time of sampling. The Charlson comorbidity index was similar in the two groups, because age was matched and the underlying conditions did not differ significantly. All MILD patients recovered fully without sequelae, while 4 SEVERE patients required extracorporeal membrane oxygenation. One patient died of persistent pneumonia and septic shock. Among the laboratory parameters, hypoalbuminemia, high neutrophil-to-lymphocyte ratio, and increased serum C-reactive protein and lactate dehydrogenase levels were associated with disease severity (Table 1).
In this study, we first assessed the profiles of immunological determinants depending on disease severity in Korean COVID-19. To examine the immune-related transcriptome profiles induced by COVID-19 infection, we performed nCounter Human Immunology gene expression assays for PBMCs from 20 HC, 8 SEVERE, and 20 MILD samples (Fig. 1A). Using PCA, we found that HC was clearly separated from both SEVERE and MILD, while the two patient groups intermingled (Fig. 1B). These data imply that altered expression of immune-related genes is a transcriptional hallmark of COVID-19 and that the overall immune transcriptome profiles are similar in SEVERE and MILD groups in Korea. To determine which genes are differentially expressed in COVID-19 patients, we compared the expression of immune-related genes between HC and COVID-19 patients. In all, 298 DEiGs were identified, and they were mainly involved in the cytokine–cytokine receptor interaction and NF-κB signaling pathways (Fig. 1C and D). The same analysis was repeated for each patient group separately, and we identified 230 and 255 DEiGs in the SEVERE and MILD samples, respectively (Supplementary Fig. 1A and C). The cytokine–cytokine receptor interaction was the top enriched pathway in both the SEVERE and MILD groups (Supplementary Fig. 1B and D).
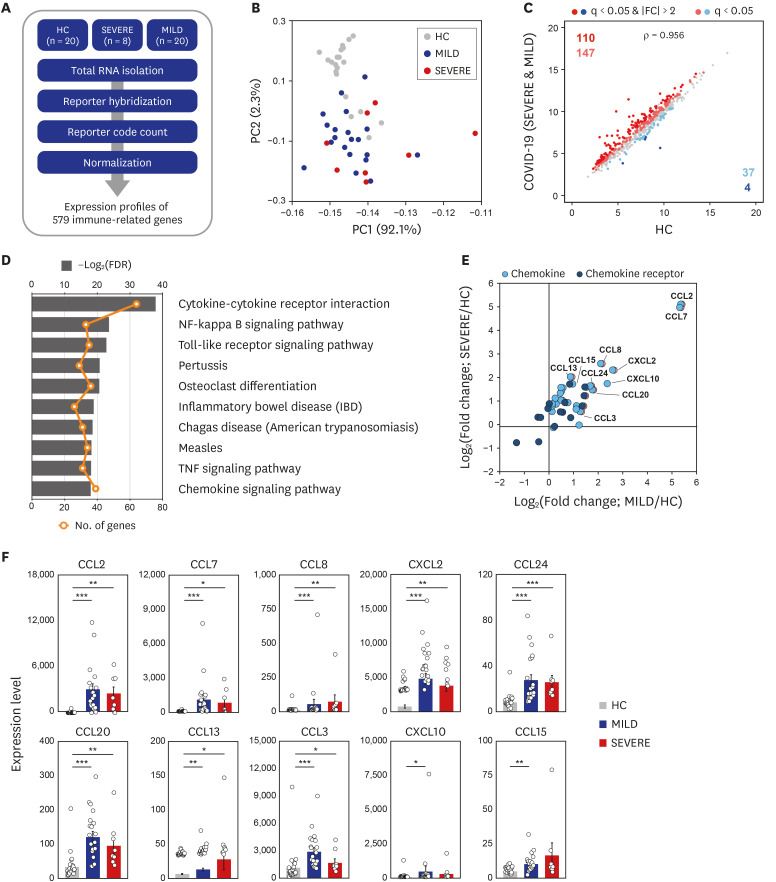
Fig. 1
Transcriptome analysis reveals that immune gene expression profiles of COVID-19 patients are distinct to HC. (A) Schematic diagram of the immune transcriptome analysis in this study. (B) A result of principal component analysis of log2-transformed 579 immune gene expression levels. (C) The scatter plots representing 579 immune genes with the log2-transformed FPKM for COVID-19 patients compared to HC. (D) The ten most significantly enriched KEGG pathways of the 298 DEiGs from COVID-19 patients compared to HC. (E) Log2-transformed fold changes of chemokine and chemokine receptor genes from MILD (x-axis) and SEVERE (y-axis) vs. HC. (F) Expression levels (FPKM) of marked chemokines in (E). Error bar indicates standard error of mean. P values were calculated using Mann-Whitney U test and adjusted P values (FDR) were shown.
COVID-19 = coronavirus disease 2019, HC = healthy controls, FPKM = fragments per kilobase exon-model per million reads mapped, KEGG = Kyoto Encyclopedia of Genes and Genomes, DEiG = differentially expressed immune gene, MILD = mild/moderate, SEVERE = severe/critical, FDR = false discovery rate, IBD = inflammatory bowel disease, TNF = tumor necrosis factor, CCL = C-C motif chemokine ligand, CXCL = C-X-C motif chemokine ligand.
*P < 0.05; **P < 0.01; ***P < 0.001.
![]()
We then investigated the full list of gene families associated with the cytokine–cytokine receptor interaction pathway. Using the HUGO gene nomenclature database, members of several gene families were identified, including chemokines, ILs, TNFs, and IFNs (Supplementary Fig. 2A). Notably, C-C motif (CC) chemokines (CC chemokine ligand [CCL] 2, CCL7, CCL8, CCL24, CCL20, CCL13, and CCL3), C-X-C motif (CXC) chemokines (CXC chemokine ligand [CXCL] 2 and CXCL10), and chemokine receptor subfamilies were most numerous, and were significantly (FDR < 0.05) upregulated in both MILD and SEVERE COVID-19 patient groups (Fig. 1E and F; Supplementary Fig. 2B). Similar upregulated gene expression patterns were observed in the other three family members including ILs, IFNs, and TNFs (Supplementary Fig. 3). It was noted that IL7R and CD40LG levels were significantly depressed in SEVERE patients, compared with HC (Supplementary Fig. 3). Together, these data suggest that abnormal inflammatory chemokine generation represents as common immune features during COVID-19.
Elucidation of molecular signaling pathways of immune transcriptome during COVID-19
Next, we explored the second top hit pathway, NF-κB signaling (Fig. 1D), which is one of the major hyper-activated pathways following COVID-19 infection.21 With some exceptions (such as LCK, CD40LG, PLAU, PTGS2, and TRAF5), the expression of TLR4 and its related/downstream signaling molecules (CD14, myeloid differentiation primary-response 88 [MYD88], IRAK1, TRAF6, TIRAP, and TICAM) were significantly (FDR < 0.05) upregulated (Fig. 2A). In addition, most NF-κB signaling pathway genes (NFKBIA, NFKB1, RELA, and NFKB2) were significantly (FDR < 0.05) upregulated (Fig. 2A). These data suggest that TLR4-mediated NF-κB signaling pathway activation is involved in the upregulation of inflammatory responses in patients with COVID-19 infection.
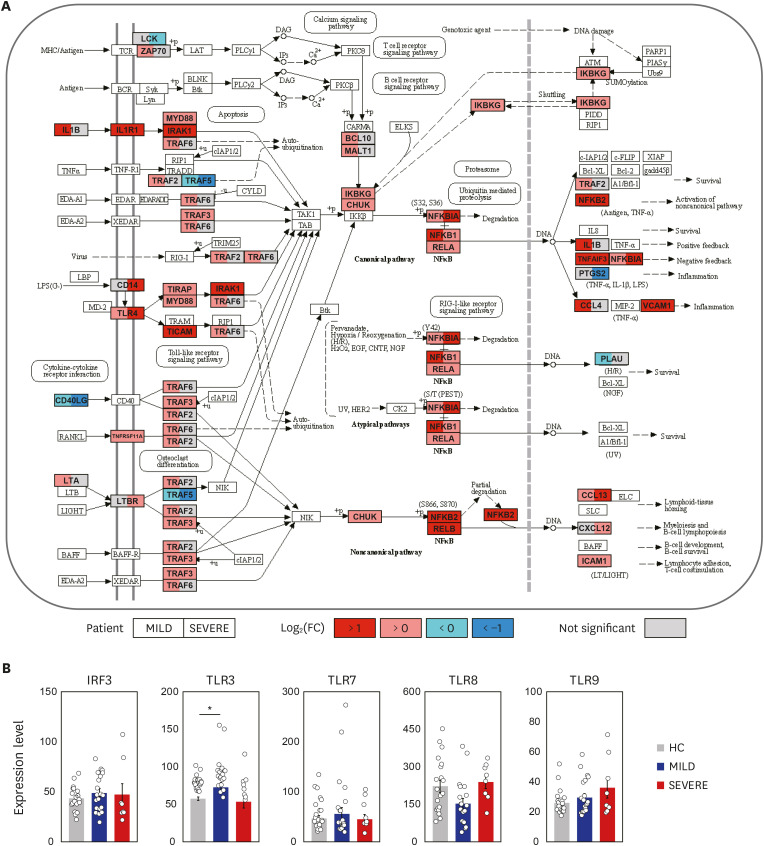
Fig. 2
COVID-19 infection boosts NF-κB signaling pathway. (A) The left (MILD) and right (SEVERE) sides of box represent the mean fold change in mRNA levels, compared with HC. The NF-κB signaling pathway was adopted from KEGG database (accession number: hsa04064). (B) The expression levels of IRF3, TLR3, TLR7, TLR8 and TLR9 were represented by FPKM. Error bar indicates standard error of mean. P values were calculated using Mann-Whitney U test and adjusted P values (FDR) were shown.
COVID-19 = coronavirus disease 2019, NF = nuclear factor, MILD = mild/moderate, SEVERE = severe/critical, HC = healthy controls, KEGG = Kyoto Encyclopedia of Genes and Genomes, FPKM = fragments per kilobase exon-model per million reads mapped, FDR = false discovery rate, TLR = toll-like receptor, IRF = interferon regulatory factor.
*P < 0.05.
![]()
Interestingly, there were no significant differences in the expression of IFN regulatory factor (IRF) 3, TLR3, TLR7, TLR8, and TLR9, all of which are related to putative viral signaling,2223 between COVID-19 patients and HC (Fig. 2B). We also found that IL1B and its downstream inflammatory signaling molecules (IL1R1, MYD88, IRAK1, TRAF6, NFKBIA, NFKB1, and RELA) were dramatically elevated in COVID-19 patients (Fig. 2A). The data suggest that the upregulated profiles of TLR4, IL1R, and NF-κB signaling pathway molecules in COVID-19 patients are presumably associated with the altered immune responses to viral components, host damage-associated molecular pattern (DAMP) signals, or cytokine signaling activation,10 and may contribute to uncontrolled pathological inflammation.
Identification of SEVERE-specific immune target genes
To understand the pathophysiological differences between SEVERE and MILD patients better, we then examined whether there are any transcriptomic differences between the two COVID-19 patient groups. Directly comparing the gene expression profiles between the SEVERE and MILD groups, no genes were significantly (FDR < 0.05) differentially expressed, which might have been partly hindered by the heterogeneity of the presentation of disease severity. Therefore, we performed two pairwise transcriptome comparisons: SEVERE vs. HC and MILD vs. HC. From these comparisons, 58 DEiGs were identified showing significant (FDR < 0.05) changes in gene expression between SEVERE and HC, which are potential therapeutic targets, but none between MILD and HC (Fig. 3A). Intriguingly, the highly expressed SEVERE-specific upregulated genes were mainly associated with complement activation (C9 and C1QA), autoimmunity (AIRE and PRKCD), and inflammatory processes (CXCL11, CCL16, and S100A9) (Fig. 3B). The SEVERE-specific downregulated genes were linked to major histocompatibility complex proteins (HLA-DRA and HLA-DMA), T-cell factor/lymphoid enhancer-binding factor family (TCF7 and LEF1), and natural killer cell functions (KLRB1, KLRG1, CD160, and GZMK) (Fig. 3C).
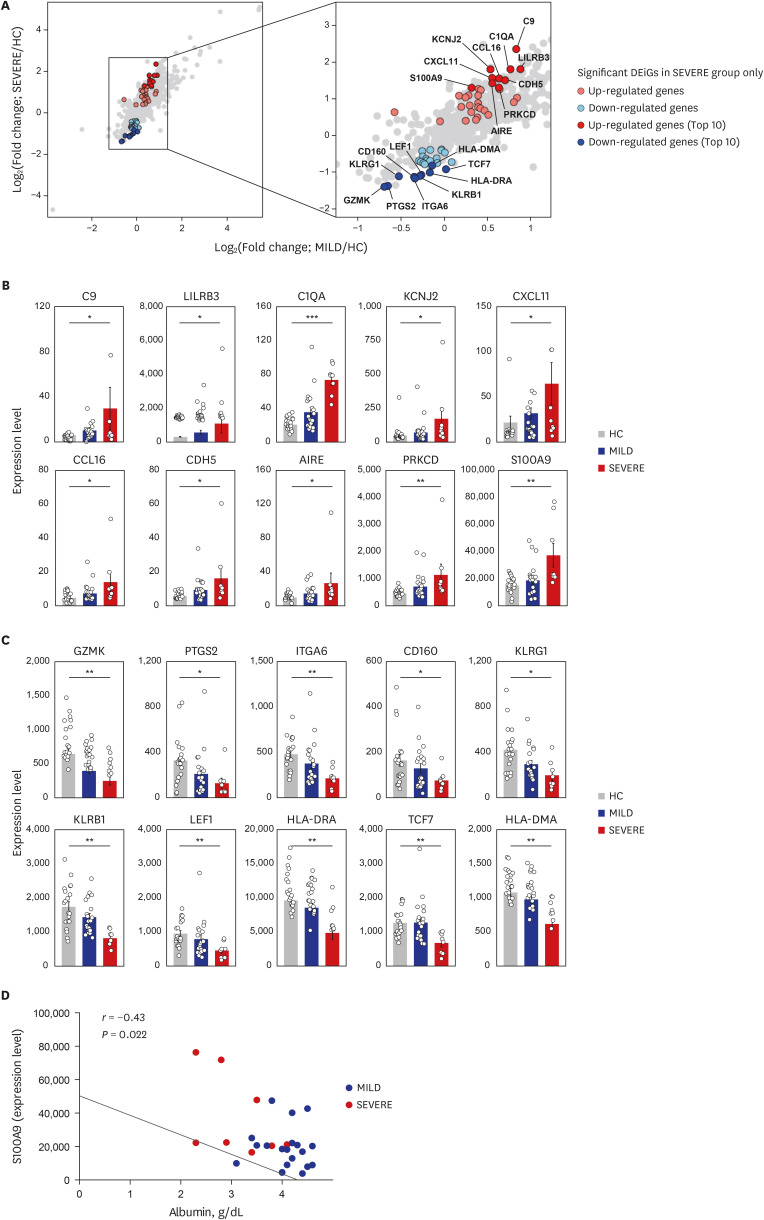
Fig. 3
Top 10 most significantly up- and down-regulated DEiGs in SEVERE patients. (A) Log2-transformed fold changes of 579 immune-genes from MILD vs. HC (x-axis) and SEVERE vs. HC (y-axis). The genes for red and blue colors indicate up- and down-regulated DEiGs in SEVERE patients, respectively. (B, C) Comparisons of expression levels of top 10 up- (B) and down- (C) regulated DEiGs. The expression level was represented by FPKM. (D) Correlation analysis between S100A9 expression level and serum albumin in MILD and SEVERE patients. Error bar indicates standard error of mean. P values were calculated using Mann-Whitney U test and adjusted P values (FDR) were shown (B, C) and Spearman's correlation is shown (D).
DEiG = differentially expressed immune gene, MILD = mild/moderate, SEVERE = severe/critical, HC = healthy controls, FPKM = fragments per kilobase exon-model per million reads mapped, FDR = false discovery rate, CCL = C-C motif chemokine ligand, CXCL = C-X-C motif chemokine ligand.
*P < 0.05; **P < 0.01; ***P < 0.001.
![]()
We then evaluated the correlation between the immune mediators and clinical parameters (Supplementary Fig. 4) and among the immune markers (Supplementary Fig. 5) in COVID-19 patients. On examining the relationship between immune markers and clinical parameters (Supplementary Fig. 4), we identified S100A9 as an important biomarker that was inversely correlated with serum albumin level in the SEVERE group (Fig. 3D).
Inflammatory signaling activation triggered by SARS-CoV-2 proteins
To gain more insight into the effects of viral components on the inflammatory responses in human PBMCs and MDMs, we then assessed whether recombinant proteins of SARS-CoV-2 (NC, S2 ECD, S1, and RBD) (Fig. 4A) induced the expression of pro-inflammatory cytokines/chemokines and NF-κB signaling activation. Notably, our data showed that recombinant NC and S2 ECD, but not the S1 subunit or RBD domain of S proteins, of SARS-CoV-2 significantly increased pro-inflammatory cytokines/chemokines in human primary PBMCs (Fig. 4B) and MDMs (Supplementary Fig. 6A and B). However, none of the proteins induced pro-inflammatory cytokine or chemokine gene expression in A549 airway epithelial cells (Supplementary Fig. 6C). In addition, either NC or S2 significantly triggered expression of various chemokines, IFNG, and S100A8/A9 in human PBMCs (Fig. 4C) and MDMs (Supplementary Fig. 6A and B). It was striking that SARS-CoV-2 NC and S2 proteins markedly increased the production of pro-inflammatory cytokine IL-6 in human PBMCs (Fig. 4D and Supplementary Fig. 6D; at 6 and 18 hours, respectively).
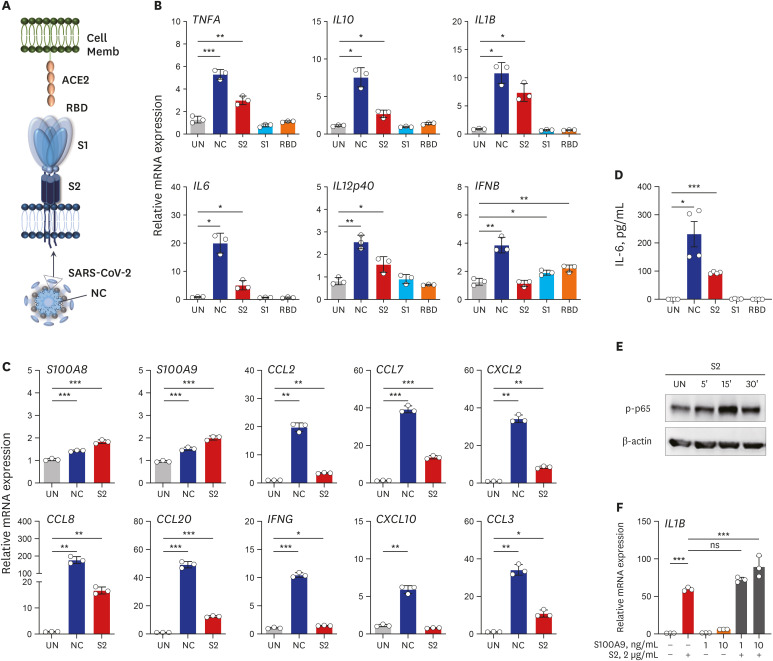
Fig. 4
Recombinant NC and S2 ECD proteins of SARS-CoV-2 robustly induce the expression of pro-inflammatory cytokines/chemokines in human primary PBMCs. (A) Schematic diagram of interaction between host cell and SARS-CoV-2 antigen. (B, C) RT-qPCR analysis of indicated genes in human primary PBMCs treated with recombinant NC, S2 ECD, S1 subunit, or RBD antigen (B) and NC or S2 ECD (C) (2 μg/mL each; for 6 hours). (D) Level of IL-6 in cell supernatant from (B) measured by ELISA. (E) Immunoblot analysis of phospho-p65 (NF-κB) in human primary PBMCs treated with S2 ECD (2 μg/mL) for indicated time. (F) RT-qPCR analysis of IL1B in human primary PBMCs treated with recombinant S2 ECD in presence or absence of indicated doses of S100A9 for 6 hours. Welch's t-test (B-D) and One-way analysis of variance (F) were used to measure the significance. Values are mean ± standard deviation. from a representative of two independent experiments performed in triplicate (B, C, F) or mean ± standard error of mean. of pooled data from two independent experiments (D).
NC = nucleocapsid, S = spike, ECD = extracellular domain, SARS-CoV-2 = severe acute respiratory syndrome coronavirus-2, PBMC = peripheral blood mononuclear cell, RT-qPCR = real-time quantitative polymerase chain reaction, RBD = receptor binding domain, IL = interleukin, ELISA = enzyme-linked immunosorbent assay, NF = nuclear factor, IFN = interferon, CCL = C-C motif chemokine ligand, CXCL = C-X-C motif chemokine ligand, RBD = receptor binding domain, UN = untreated.
*P < 0.05; **P < 0.01; ***P < 0.001.
![]()
In addition, S2 protein triggered NF-κB signaling activation in human PBMCs within 30 minutes (Fig. 4E). Furthermore, S2 protein in presence of recombinant S100A9 significantly induced the IL1B mRNA expression in PBMCs, as compared to those stimulated with either S2 protein or S100A9 alone (Fig. 4F). These data strongly suggest that the NC and spike protein S2 ECD can trigger inflammatory responses and NF-κB signaling activation, and S100A9 may act as a mediator in a positive feedforward loop of inflammatory signaling activation in human PBMCs and MDMs. Together, these data demonstrated for the first time that SARS-CoV-2 proteins NC and S2 ECD trigger the activation of inflammatory cytokine/chemokine responses in human PBMCs and MDMs.
DISCUSSION
We found that many CC chemokines, ILs, and type I IFNs are highly upregulated in PBMCs from both MILD and SEVERE patients, compared with those from HC. Results in this study partially correlate with recent reports that SEVERE cases are associated with defective immune responses, i.e., lymphopenia, high neutrophil-to-lymphocyte ratio, and increased inflammatory cytokine levels.1524252627 We also have similar data with recent studies from Wuhan, China, that found excessive expression of chemokines (CCL2/MCP1, CXCL10/IP10, CCL3/MIP1A, and CCL4/MIP1B) in bronchoalveolar lavage fluid and PBMCs from patients with SARS-CoV-2.28 Huang et al.17 showed that intensive care unit patients with clinical complications had high levels of pro-inflammatory cytokines and chemokines, including IL-2, IL-7, IL-10, granulocyte colony-stimulating factor, CXCL10, CCL2, CCL3, and TNF-α. In addition, our data partly correlate with recent studies reported by Lee et al.29 that Korean patients with COVID-19 had hyper-inflammatory phenotypes with TNF/IL-1β upregulation in all types of cells among PBMCs. In that study, severe patients exhibited co-existed pattern of type I IFN and TNF/IL-1β responses in monocytes.29 The discrepancy from previous studies29 and ours might be due to different subjects as well as single vs. total cell population in PBMCs. Our study has a limitation of immune transcriptome analysis in mixed cell population of PBMCs. Future studies are requested to analyze the immune profile at a single-cell level, and in a larger population than the present data.
Innate immune responses are triggered by pattern-recognition receptors, including TLRs and nucleotide-binding oligomerization domain-like receptors, and activate the complicated intracellular signaling cascades that culminate in the activation of NF-κB pathways.30313233 TLR signaling pathways are mediated by the components, including sensors that recognize certain pathogen-associated molecular patterns (PAMPs) and DAMPs and the adaptors that transduce signals.32 Among TLRs, TLR4 can recognize lipopolysaccharide, other PAMPs, and DAMPs at the cell surface, whereas TLR3, TLR7, TLR8, and TLR9 are exclusively expressed in endosomal compartments and recognize viral components.313233 TLR4 is the only TLR to transduce innate immune signals through both MyD88 and Toll-IL-1 receptor-domain-containing adaptor-inducing IFN-β to activate NF-κB and IRF signaling, respectively.313233 NF-κB signaling pathway is required for pro-inflammatory cytokine/chemokine generation and the production of antimicrobial proteins3334; IL-1 family members are involved in the initiation of potent inflammatory responses, orchestration of innate and adaptive immunity, and development of sepsis.3536 Although both TLR and IL-1 signaling activation are critical for innate immune defense against a variety of pathogens, dysregulation of this signaling pathway can lead to pathogenesis of various diseases including inflammatory and autoimmune diseases.35363738
The increased S100A9 seems to be critically important because hypoalbuminemia is associated with disease severity in COVID-19 patients.7 S100A8/A9 (a heterodimer complex of S100A8 and S100A9 proteins)39 is a DAMP signal as a TLR4 ligand.40 The elevated expression of S100A8/A9 is induced by inflammation, and secreted S100A8/A9 further amplifies inflammatory soluble cytokines/chemokines, forming a feed-forward loop affecting the persistent inflammation.41 Importantly, S100A8/A9, as a ‘soil signal’, mediates metastasis of melanoma or breast cancers to the lung.40 Since S100A8/A9 protein is involved in the pathogenesis of numerous inflammation-associated and autoimmune diseases,4042 our findings provide new insight into the pathogenesis of COVID-19, and may contribute to therapeutic approaches based on the S100A9-CC chemokine-mediated inflammatory signaling.
Notably, we found that the recombinant NC and S2 ECD, but not the S1 subunit or RBD domain of S proteins, of SARS-CoV-2 significantly increased pro-inflammatory cytokines/chemokines in human primary PBMCs and MDMs. These data strongly suggest that the viral proteins are able to induce pro-inflammatory responses in human immune cells. Similarly, a previous study on the S protein of SARS-CoV showed that it was selectively recognized by lung surfactant protein and effectively activated macrophages, but not dendritic cells, to produce TNF-α, IL-6, and IL-8.43 However, unlike our results, authors proclaim that purified S-protein did not trigger TLR2 or TLR4 pathway due to unresponsiveness of NF-κB signaling.43 Recently, cytomegalovirus protein US31 was reported to directly interact with NF-κB2, resulting induction of NF-κB2-induced inflammation in macrophages.44 Duette et al.45 revealed that release of extracellular vesicle, which is likely to contain viral proteins, during HIV infection promoted viral replication and macrophage-mediated inflammatory responses in coordination with HIF-1α induction. These results indicate that some viral proteins function as a pro-inflammatory mediator depending on their functional structures and species origin.
Recent promising results suggest that dexamethasone has beneficial effects to reduce deaths of patients receiving invasive ventilation or oxygen.46 Our findings provide a rationale to use dexamethasone, a ligand for glucocorticoid receptor, which interferes with TLR-dependent inflammatory signaling through multiple mechanisms.4748 Indeed, it has been long suggested that the blockade of TLR signaling through molecular checkpoints may contribute to developing the potential treatment against specific infections and/or other diseases.49 Taken together, these data provide novel insights into the idea that the amelioration of excessive TLR4-mediated innate signaling might be beneficial for treatment of COVID-19.
 XML Download
XML Download