

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Plasma cell myeloma (PCM) is a multifocal neoplastic proliferation of plasma cells in the bone marrow (BM) [1]. Karyotype abnormalities are the main drivers of PCM, including hyperdiploidy characterized by trisomies of chromosomes 3, 5, 7, 9 11, 15, 19, and 21, and rearrangements involving the immunoglobulin heavy locus (IGH) gene translocations [2].
Recent studies have expanded the genetic spectrum of PCM regarding genetic mutations. The RAS pathway is the most frequently mutated pathway in PCM, with about 20% of newly diagnosed patients with PCM having driver mutations in KRAS or NRAS [3, 4]. Mutations occur at codons 12 and 61 in KRAS and NRAS, respectively, preventing GTP hydrolysis and keeping RAS in its active state, subsequently activating the mitogen-activated protein kinase (MAPK) pathway [5]. In addition, about 5% (4% to 12%) of patients with PCM harbored the BRAF mutation at diagnosis, mostly on amino acid V600 [3, 4, 6-10]. BRAF V600E causes constitutive activation of the RAS pathway, presumably leading to increased cell growth and preventing apoptosis [11]. It has been known that activating mutations in the RAS-RAF-MAPK pathway are more frequently observed in relapsed and refractory PCM and are associated with worse prognosis, shorter patient survival, and tumor progression; however, a majority of studies have been performed before the era of novel therapeutic agents [12-15].
In this study, we analyzed the KRAS, NRAS, and BRAF genes to evaluate the prevalence of mutations in these genes in patients from a single Korean institute. We also have studied their associations with clinical characteristics and karyotypes, and evaluated their influence on clinical outcomes.
MATERIALS AND METHODS
Patients
We explored 50 patients who were newly diagnosed with PCM between 2009 and 2012 at Seoul St. Mary’s Hospital. We selected available BM samples of inpatients who were previously performed with a BM study and were treated for PCM. We analyzed the data in April 2020. This study was performed according to the Declaration of Helsinki and approval for this study was obtained from the Institutional Review Board of Seoul St. Mary’s Hospital, The Catholic University of Korea (KC12SISE0594).
KRAS and NRAS mutation analysis by pyrosequencing
Genomic DNA from BM aspirates were isolated using a Wizard Genomic DNA Purification kit (Promega, Madison, WI, USA). Pyrosequencing was carried out using a PCR primer mix for KRAS exon 2 (codon 12, 13) and NRAS exon 3 (codon 61) (Supplementary Table 1). Each PCR mix contained forward and reverse primers (1 μL), 10× PCR buffer (2 μL), dNTP (0.2 μL), water (19.65 μL), Hotstar Taq Polymerase (0.15 μL), and 2 μL of genomic DNA for a total volume of 25 μL. PCR was done on a GeneAmp PCR system 9700 Thermal Cycler (Thermo Fisher Scientific, Waltham, MA, USA) with an initial activation step at 95°C for 15 min, 45 cycles of denaturation at 95°C for 30 sec, annealing at 60°C for 30 sec, and extension at 72°C for 30 sec, followed by a final extension cycle at 72°C for 15 min. Pyrosequencing was performed with an 80 μL final volume, containing 40 μL of biotinylated PCR product with high purity water, 37 μL of PyroMark Binding Buffer, and 3 μL of streptavidin beads (GE Healthcare, Uppsala, Sweden). Pyrosequencing was performed using a PyroMark Q96 ID instrument according to the manufacturer’s instructions (Biotage, Uppsala, Sweden).
BRAF V600E detection by AS-PCR
We performed additional BRAF V600E detection tests using a Real-Q BRAF V600E detection kit (BioSewoom Inc., Seoul, Korea) on an Applied Biosystems 7500 Real-time PCR (Thermo Fisher Scientific) according to the manufacturer’s instructions. Briefly, PCR was performed in a 25 μL reaction volume containing 10 μL DNA, 12.5 μL 2× PCR reaction mix, and 2.5 μL BRAF probe and primer mixture. Reactions were performed for 40 cycles of 50°C for 2 min, 95°C for 10 min, 95°C for 15 sec and 58°C for 45 sec. The assay was repeated at least two times.
Definitions and Statistics
Comparisons of clinical and laboratory parameters at diagnosis between patient subgroups were done with the Mann-Whitney test or Fisher’s exact test, respectively. Stages were classified according to the International Staging System for multiple myeloma [16]. Treatment response was evaluated according to the International Myeloma Working Group (IMWG) criteria [17]. Events for progression-free survival (PFS) were indicated as the first progression after frontline treatment or any cause of death. Overall survival (OS) was indicated as the time from initiation of frontline treatment to death (from any cause) or the date of the last follow-up. OS and PFS were determined using the Kaplan-Meier method and compared using a log-rank test. Variables with P<0.1 in univariate analyses were entered into multivariate models using Cox proportional hazards regressions with a backward stepwise model selection. P-values <0.05 were considered significant. All statistical analyses were conducted using R.3.1.1 statistical software (http://cran.r-project.org/).
RESULTS
Clinical characteristics of patients
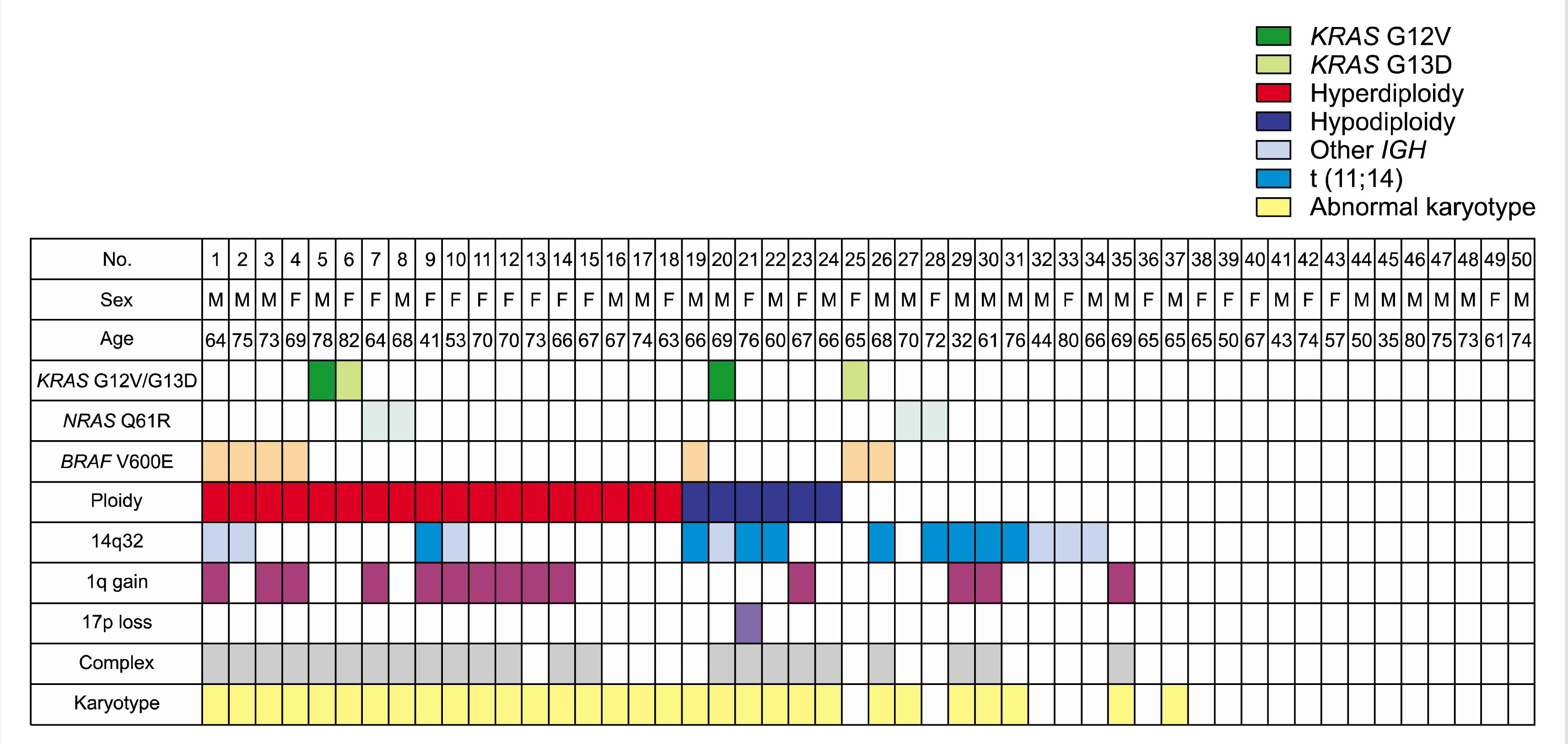
The patient demographics, clinical, and laboratory characteristics are summarized in Table 1. The median age of patients was 66 years (range, 31–82). The most frequent type of myeloma was IgG (54%, 27/50), followed by IgA (20%), light chain (18%), IgD (6%), and IgM (2%). Karyotype analyses and interphase FISH were performed using diagnostic BM aspirates with the same methods used in a previous study [18]. Results were classified and described according to the 2016 International System for Human Cytogenetic Nomenclature (ISCN) guidelines [19]. Of the 50 patients, 31 (62%) harbored abnormal karyotypes. Hyperdiploidy (≥47 chromosomes) and hypodiploidy (≤45 chromosomes) were observed in 18 (36%) and 6 patients (12%), respectively. Sixteen patients presented with IGH gene rearrangements including t(11;14) (N=9), t(8;14) (N=1), and other IGH gene rearrangements (N=6). Fourteen patients (28%) showed 1q gain and one patient (2%) had a 17p deletion. Complex karyotype, defined as ≥3 chromosome abnormalities, was identified in 23 patients (46%). Regarding the selection of treatment, we had intention-to-treat with autologous stem cell transplantation following frontline chemotherapy for 14 patients (28%), whereas 36 patients (72%) classified as transplant-ineligible received chemotherapy consisting of bortezomib-melphalan-prednisolone for their frontline treatment.
Table 1
Baseline characteristics of the patients.
![]()
Activating mutations in RAS-RAF-MAPK pathway
RAS mutations were detected in 16% patients (8/50), including four KRAS mutations and four NRAS mutations. BRAF mutations were detected in seven patients (14%). KRAS and NRAS mutations were mutually exclusive. A patient with both KRAS G13D and BRAF mutations was found (Fig. 1). Thus, fourteen patients showed any KRAS, NRAS, and/or BRAF (RAS/RAF) mutations. RAS/RAF mutations were more commonly identified in patients with abnormal karyotype [odds ratio (OR), 5.368; 95% CI, 1.048–27.502; P=0.050] and complex karyotype (OR, 4.423; 95% CI, 1.153–16.964; P=0.031). Other parameters included age, sex, type of monoclonal proteins, hemoglobin, creatinine, total protein, albumin, LDH, and β2-microglobulin. The stage according to the International Staging System and frontline treatment type were distributed equally or non-significantly different according to RAS/RAF mutations.
In the total cohort, there was an overall response rate of 98% (49 of 50 patients) through frontline treatment: 23 patients (46%) with complete response, 13 patients (26%) with very good partial response, and 13 patients (26%) with partial response. The remaining patient showed stable disease despite 6 cycles of treatment with bortezomib-melphalan-prednisolone, finishing her frontline treatment early due to unacceptable peripheral neuropathy. RAS/RAF mutations did not provide a significant impact on either complete response rate or the achievement of partial response of any degree (Table 2).
Table 2
Characteristics of patients with any KRAS, NRAS, and/or BRAF mutations.
![]()
With a respective median PFS of 23.5 months (95% CI, 16.5–25.6) and median OS of 105.7 months (95% CI, 63.7–not estimable) (Supplementary Fig. 1), RAS/RAF mutations were significantly associated with poor median PFS [24.0 mo (95% CI, 16.5–49.3) vs. 18.2 mo (3.6–24.2), P=0.015, Fig. 2A]. There was no statistical difference in median OS according to the presence of RAS/RAF mutations (85.2 mo vs. not estimable, Fig. 2B). In the subgroup analysis for PFS, BRAF V600E mutations had a significantly negative impact on median PFS [18.2 mo (95% CI, 1.8–24.2) for patients with BRAF V600E mutation (N=7) vs. 23.9 mo (95% CI, 16.5–31.2 mo) for patients with wild-type BRAF (N=43), P=0.04, Fig. 2C]. Patients with RAS mutations, presented in either KRAS and NRAS (N=8), showed a statistical trend of poor median PFS compared to those without RAS mutations (N=42) [16.3 mo (95% CI, 0.4–26) vs. 23.9 mo (95% CI, 17.6–29), P=0.081, Fig. 2D]. Multivariable analysis showed that RAS/RAF mutations were independent factors associated with poor PFS (hazard ratio=2.28, 95% CI, 1.15–4.5, P=0.018). However, the factor of BRAF V600E mutation lost their statistical significance in the multivariable analysis (Table 3, Supplementary Table 2).
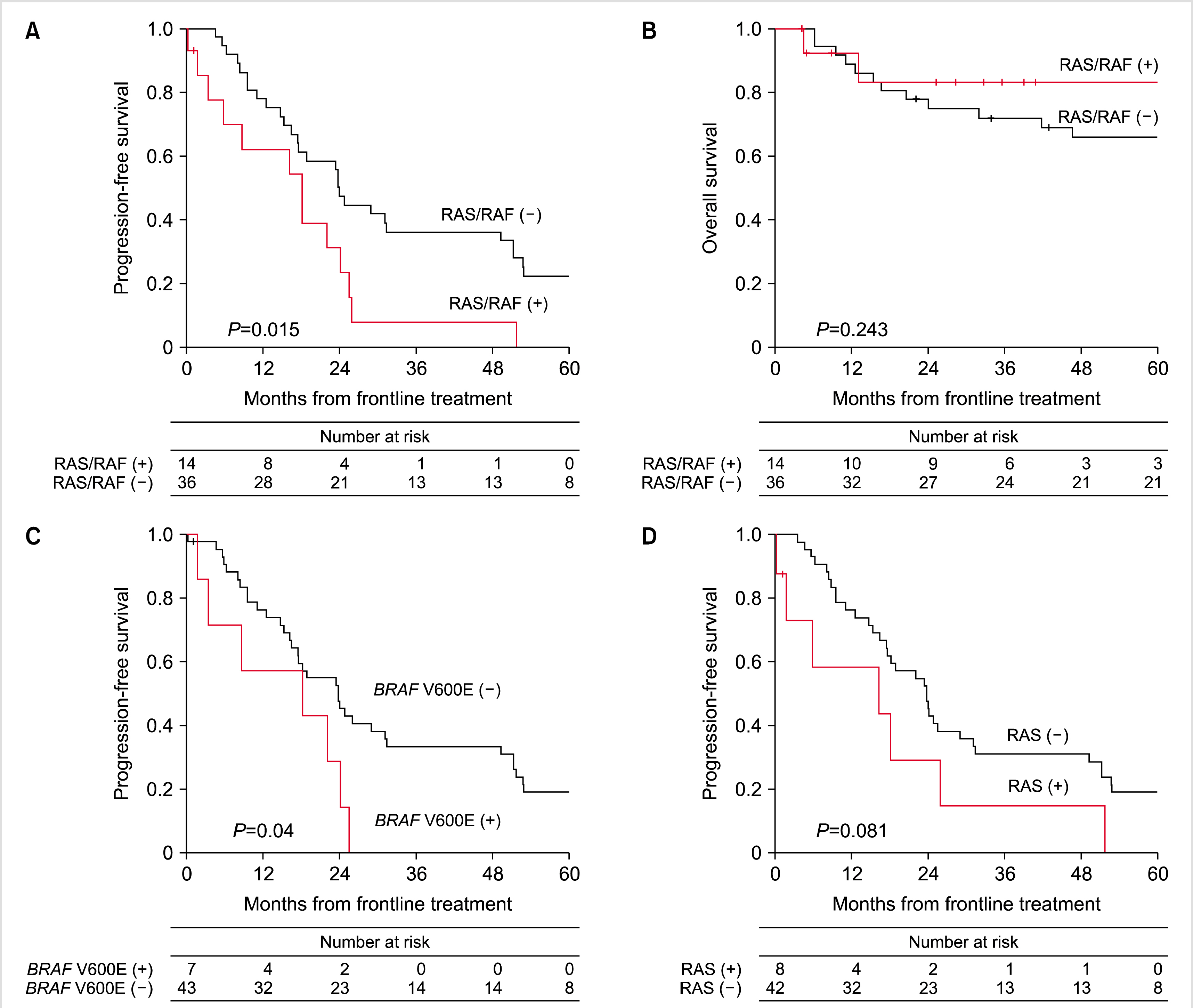
Fig. 2
linical outcomes of patients with any KRAS, NRAS, and/or BRAF mutation [RAS/RAF(+)] or without mutations [RAS/RAF(-)] (A, B). Comparison between subgroups; BRAF V600E (+) vs. BRAF V600E (-) (C) and RAS (+) vs. RAS (-) (D).
![]()
Table 3
Univariable and multivariable analysis for progression-free survival.
![]()
DISCUSSION
In this study, we identified 14 patients with PCM and RAS/RAF mutations (28%). KRAS and NRAS mutations were detected in 16% of patients, and BRAF V600E was in 14%. The prevalence was similar with the results from previous studies except for BRAF V600E, which was slightly higher than others [4, 6-9, 20]. This may be explained by the higher sensitivity of AS-PCR in detecting BRAF V600E compared to other molecular techniques, including pyrosequencing [21]. We also found that RAS mutations were mutually exclusive, but not with BRAF V600E [22]. Interestingly, RAS/RAF mutations were associated with abnormal karyotype and complex karyotype. These results were in line with previous findings that RAS mutations can cause a transition from the pre-malignant monoclonal gammopathy of undetermined significance to PCM [23, 24]. However, RAS mutations did not correlate with clinical stage, such as ISS [25].
From a clinical perspective, the prognostic significance of RAS/RAF mutations in PCM is controversial. The application of novel therapeutic agents has changed the influence of these mutations. One study showed that RAS mutations appeared to be significantly associated with a favorable outcome [26]. Another study showed that functional activation of the RAS pathway was observed in 75% of patients with relapsed/refractory PCM and about half had RAS/RAF mutations [27]. In this study, we demonstrated that RAS/RAF mutations were associated with poor outcomes, such as a shorter PFS. Aside from RAS/RAF mutations, a subgroup with BRAF V600E mutations also provided poor PFS compared to the wild-type RAS/RAF. Because the activation of the MAPK pathway via RAS/RAF mutations was concordant with the gene expression profile data of the same patients, inhibition of this signaling would be effective in this subgroup of patients [7, 27, 28]. Of them, BRAF has received considerable attention as a result of the success of targeted malignant melanoma therapy [29]. The genomic and transcriptional mutant allele burdens of BRAF were highly concordant in patients with BRAF-mutant PCM [9]. These findings further strengthen the hypothesis that patients with BRAF V600E mutations may benefit from new targeted treatments with BRAF inhibitors, such as vemurafenib [30] and dabrafenib [31]. A recent study suggested that the development of mutation-specific KRAS inhibitors could be of great value in patients with KRAS-mutant PCM [32, 33]. Moreover, results of the correlation between RAS/RAF mutations and complex karyotypes suggested the therapeutic benefit of checkpoint inhibitors in this group [34].
Our study has several limitations. This is a retrospective study using selected BM samples of inpatients considered to be in a more severe condition with a high disease burden. Thus, the proportion of karyotype abnormalities and ISS stage II were relatively higher compared to a previous study in Asian patients [35]. There is also a possibility that the prevalence of KRAS, NRAS, and BRAF mutations have been overestimated due to the same reason. Next, the number of enrolled samples was small and their baseline characteristics, including frontline treatment, were heterogeneous. Our results should be further validated with a large number of cases before implementation in the clinic.
Consequently, this study first showed the frequency of RAS/RAF mutations in Korean patients with PCM. Screening of these mutations could be considered as a routine clinical test at the time of diagnosis and follow-up due to their influence on clinical outcomes and their potential as a therapeutic target.
 XML Download
XML Download