

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Lung cancer is obviously the first cause of cancer-related mortality in the world. Its five-year survival rate is under 15% [1]. About 70% of lung carcinoma patients present with metastatic diseases at the time of diagnosis, contributing to a high mortality of lung cancer [2]. It has been reported that patients with lung tumor in advanced stages will die within 18 months after diagnosis [3]. Lung cancer is categorized into two histologic classes: non-small cell lung cancer (NSCLC) that constitutes approximately 80%–85% of all lung cancer cases and small-cell lung cancer (SCLC) [2]. Although significant achievement has been made over the past decade with treatment of NSCLC using epithelial growth factor receptor (EGFR) targeted therapies, angiogenesis inhibitors, and immune checkpoint inhibitors, patients with lung cancer still suffer from poor prognosis and aggressive progression of the disease [24]. Therefore, it is crucial to find novel drugs and develop more efficient therapeutic strategies for treating NSCLC.
Autophagy is a cellular process to remove and recycle dysfunctional cytoplasmic components. During the autophagy process, damaged organelles or misfolded proteins in the cytoplasm are sequestered by large double-membrane vesicles called autophagosomes. Autophagosomes are finally fused with lysosomes to form autolysosomes where sequestered cytoplasmic constituents are degraded by lysosomal enzymes [5]. Autophagy occurs in most tissues as a cytoplasmic quality-control mechanism to maintain intracellular homeostasis. However, autophagy can be triggered by various forms of intracellular stress such as nutrient depletion, reactive oxygen species (ROS) generation, hypoxia, and DNA damage [6]. Additionally, autophagy is implicated in numerous human diseases, including neurodegenerative disorders, liver diseases, muscular diseases, pathogen infection, and cancers [7]. The role of autophagy in cancer progression is dependent on tumor type, pathological stage, and clinical stages. In general, autophagy inhibits tumorigenesis by eliminating genome instability and tissue damage in early stages. However, it also promotes tumor growth and chemoresistance in advanced stages of cancer [89]. Such context-dependent roles of autophagy in cancer have driven clinical interventions to induce or inhibit autophagy as cancer therapies [10].
AMP-activated protein kinase (AMPK) is a well-recognized serine/threonine protein kinase that regulates metabolic pathways in response to cellular stresses and energy deprivation [11]. It has been reported that activation of AMPK pathway plays a pivotal role in autophagy process. When intracellular ATP is exhausted and the level of AMP is increased, AMPK undergoes a conformational change that allows LKB1, an upstream kinase, to easily access and activate AMPK [12]. Activated AMPK then phosphorylates various substrates and transcription factors to modulate metabolism. Among various substrates of AMPK, UNC-51-like kinase1 (ULK1) is known to promote autophagy under glucose starvation [13]. As downstream molecules, regulatory associated protein of mammalian target of rapamycin (mTOR) (Raptor) and tuberous sclerosis complex 2 can deactivate mammalian target of rapamycin complex 1 (mTORC1) which inhibits ULK1 by blocking the interaction between AMPK and ULK1 [1415]. Therefore, AMPK triggers autophagy not only by directly activating ULK1, but also by disrupting the suppressive effect of mTORC1 on ULK1. Numerous studies have demonstrated that the AMPK/mTOR signaling axis plays a vital role in the process of autophagy [1113].
Morusin, a marker constituent of Morus alba L., has been traditionally used for lung diseases such as cough, asthma, and pulmonary congestion in Korea. Morusin exhibits various biological activities including anti-oxidant, anti-convulsant, anti-microbial, anti-inflammatory, and anti-cancer effects [16171819]. Especially, morusin can trigger apoptosis in NSCLC cells by suppressing EGFR activity [20]. The autophagy inducing activity of morusin has been reported in human ovarian cancer cells [21], but not in NSCLC cells. Interestingly, we have previously found that methylene chloride extract of Morus alba L. can induce autophagy in NSCLC cells, although the precise mechanism and the active compound have not been determined yet [22]. Thus, the objective of the current study was to investigate whether morusin could trigger autophagy in NSCLC cells and explore the underlying mechanism. We also examined the role of morusin-induced autophagy in cancer cell survival to provide some insight into controversial roles of autophagy in cancer progression.
Go to : 
SUBJECTS AND METHODS
Reagents and antibodies
Morusin was purchased from ChemFaces (Wuhan, China) and dissolved in dimethyl sulfoxide (DMSO) at 100 mM as a stock solution. MTT [3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide] was bought from Duchefa (Haarlem, The Netherlands). Trypan blue was purchased from WelGENE (Daegu, Korea). Compound C, 4,6-diamidino-2-phenylindole (DAPI), DMSO, propidium iodide (PI), RNase A, and bafilomycin A1 were obtained from Sigma-Aldrich (St. Louis, MO, USA). Primary antibodies against autophagy-related 5 (Atg5), Beclin-1, LC3, phospho-AMPK (Thr172), AMPK, phospho-acetyl-coA carboxylase (ACC; Ser79), ACC, phospho-mTOR (Ser2448), and mTOR were purchased from Cell Signaling Technology (Beverly, MA, USA). Primary antibodies against Atg12, phospho-p70S6 kinase (p70S6K, Ser434), p70S6K, and actin were bought from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Goat anti-mouse (Bethyl Laboratories, Montgomery, TX, USA) and goat anti-rabbit antibodies (Enzo Life Sciences, Farmingdale, NY, USA) were used as secondary antibodies.
Cell lines and cell culture
H1299 and H460 human lung carcinoma cell lines were bought from the American Type Culture Collection (ATCC, Manassas, VA, USA). They were cultured with RPMI 1640 medium (WelGENE) supplemented with 10% FBS (WelGENE) and 1% penicillin-streptomycin solution (WelGENE). Cells were maintained in a humidified incubator at 37°C with 5% CO2.
MTT assay
H460 cells were seeded at a density of 2.5 × 103 cells/well into 96-well plates and stabilized overnight. Cells were treated with indicated drugs for 72 h. Then 10 μL of MTT stock solution (4 mg/mL) was then added to the culture medium (100 μL) to make a final concentration of 0.4 mg/mL. After 2 h of incubation at 37°C, media were completely aspirated and 100 μL of DMSO was added to each well. After 96-well plates were shaken gently for 30 min to completely dissolve formazan, absorbance at 540 nm was then measured with a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Trypan blue exclusion assay
H460 cells were seeded into 12-well plates at a density of 2 × 104 cells/well and stabilized overnight. Attached cells were treated with indicated drugs. After 72 h of incubation, cells were collected. After 25 μL of cell suspension was mixed with 25 μL of 0.4% trypan blue solution, the number of unstained cells (viable cells) was counted under a microscope (Leica, Wetzlar, Germany) using a hemocytometer.
Immunofluorescence
H460 or H1299 cells were seeded onto coverslips in 6-well plates and stabilized overnight. After treatment with morusin (20 μM) for 6 h or 48 h, cells attached to coverslips were rinsed with cold phosphate-buffered saline (PBS) repeatedly and fixed with 100% methanol for 10 min at -20 °C. Subsequent, a blocking step was conducted with 3% bovine serum albumin (BSA; GenDEPOT, Katy, TX, USA) solution for 50 min at 4˚C. After washing with cold PBS, cells were incubated with primary antibody against LC3 overnight at 4°C and subsequently incubated with Alexa Fluor 488 anti-rabbit secondary antibody (Invitrogen, Carlsbad, CA, USA) for 50 min at room temperature. Cells were then counterstained with DAPI. LC3 puncta and nuclei were observed with a fluorescence microscope (Leica, Wetzlar, Germany) at ×200 magnification.
Flow cytometry
H460 cells (1 × 105 cells/well) were seeded into 6-well plates and treated with indicated drugs for 72 h. To measure sub-G1 phase, cell cycle analysis was performed. Cells were pelleted and subsequently fixed with cold 80% ethanol at −20°C overnight. Then cells were rinsed with PBS and subjected to staining with PI staining solution (50 μg/mL PI in PBS containing 30 μg/mL RNase A) in the dark for 30 min. Stained cells were then pelleted by centrifugation and resuspended in 500 μL of PBS. DNA content of cells was checked with a flow cytometer (FACSCalibeur, BD Biosciences). The proportion of each phase (sub-G1, G1, S, G2/M) of cell cycle was calculated with CellQuest Pro software (version 5.1). For Annexin V-PI double staining, cells were collected, stained with Annexin V-FITC Apoptosis Detection kit I (BD Biosciences; PharMingen) according to the protocol provided by the manufacturer, and analyzed by flow cytometry. Annexin V(+) cells were determined as apoptotic cells.
Western blot
Cells were lysed with cold RIPA Lysis and Extraction Buffer (Thermo Fisher Scientific, Rockford, IL, USA) added with a protease inhibitor cocktail (Thermo Fisher Scientific) and phosphatase inhibitors (100 mM NaF and 1 mM Na3VO4). After lysis on ice for 50 min, supernatants were collected by centrifugation. Protein concentrations were evaluated using Pierce BCA Protein Assay Kit (Pierce Biotechnology, Rockford, IL, USA) according to the manufacturer's instruction. Proteins (20 μg) were then separated on 8%–13% acrylamide gels by SDS-PAGE and subsequently transferred onto polyvinyl difluoride (PVDF) membranes (Millipore, Bedford, MA, USA) for 2 h at 60 V. After blocking the membrane with 3% BSA for 30 min at room temperature, a 1:500–1:1,000 dilution of primary antibody was added to the membrane and incubated at 4°C overnight. The membrane was washed repeatedly with TBST for 50 min and subsequently probed with a secondary antibody at 1:10,000 dilution for 50 min at room temperature. Protein expression was detected using D-Plus ECL Femto System (Donginbio, Seoul, Korea) according to the manufacturer's protocol. ImageJ software (ImageJ 1.38; National Institutes of Health) was used for densitometric analysis of immunoblots.
Statistical analysis
Results are expressed as mean ± SD of data obtained from triplicate experiments. Statistical analyses were performed with a paired Student's t-test. In all cases, differences with P-value < 0.05 were considered statistically significant.
Go to :
RESULTS
Induction of autophagy by morusin in NSCLC cells
We found that many vacuoles were formed in the cytoplasm of H460 and H1299 human lung carcinoma cells after morusin treatment. Vacuoles were detected from 6 h after morusin treatment and maintained until 48 h (Fig. 1A). Previous studies have reported that autophagic vacuoles, similar to vacuoles we observed, can appear during autophagy [2324]. Thus, we investigated whether these vacuoles might be related to autophagy induction. It is well recognized that LC3II formed by a conjugation of lipid phosphatidylethanolamine to cytosolic LC3I is recruited to the autophagosome membrane during autophagy and presented as LC3 puncta [10]. As shown in Fig. 1B, morusin treatment significantly increased the formation of LC3 puncta in H460 and H1299 cells. Such LC3 puncta remained constant from early (6 h) to late time points (48 h) after morusin treatment, consistent with results shown in Fig. 1A, suggesting that observed cytoplasmic vacuoles were related to the formation of autophagosomes (Fig. 1B). These findings are consistent with results of a previous study demonstrating that morusin can induce autophagosome formation in HeLa cells at 6 h after treatment [21]. Next, we examined whether expression levels of autophagy-related proteins were changed by morusin. Beclin-1 plays key roles in the initiation of autophagosomes and Atg5-Atg12 complex expands the autophagosome membrane [10]. Our results showed that morusin enhanced expression levels of Atg5, Atg12, beclin-1, and LC3II in H460 and H1299 cells in a time-dependent manner (Fig. 1C). In addition, the accumulation of LC3II was maintained until 48 h after treatment with morusin (Fig. 1D). Taken together, our findings demonstrate that morusin can induce autophagy in NSCLC cells.
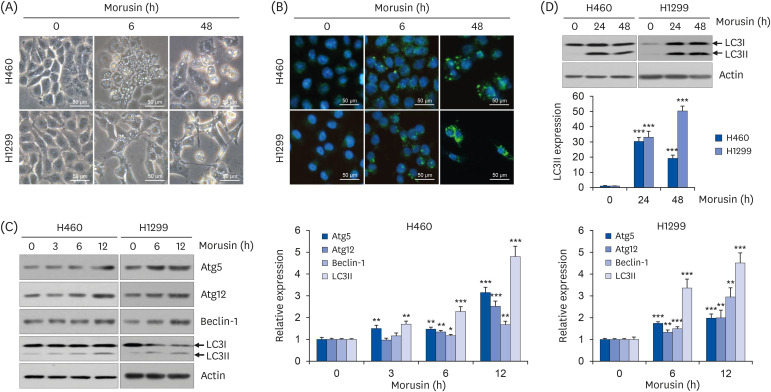 | Fig. 1Induction of autophagy by morusin in non-small cell lung cancer cells. Cells were treated with morusin (20 μM) for indicated time periods. (A) Cellular morphology was photographed with a microscope (×200 magnification). (B) Cells were subjected to immunostaining with anti-LC3 antibody and fluorescent-dye conjugated secondary antibody to observe LC3 puncta. DAPI staining was performed to visualize nuclei. LC3 puncta (green) and nuclei (blue) were photographed and merged using a fluorescence microscope (×200 magnification). (C and D) Expression of indicated protein was detected by western blot. Actin was a loading control. ImageJ software was used for densitometric analysis of immunoblots.
*P < 0.05, **P < 0.01, and ***P < 0.001 vs. untreated control group.
|
Inhibition of autophagy enhances morusin-induced apoptosis in NSCLC cells
To determine roles of morusin-induced autophagy in the survival of NSCLC cells, bafilomycin A1 known to inhibit autophagy by blocking lysosome acidification as well as autophagosome–lysosome fusion [25] was used. As shown in Fig. 2A, the expression of LC3II was increased at 12 h after treatment with morusin. It was accumulated more after co-treatment of morusin and bafilomycin A1, indicating that bafilomycin worked well (Fig. 2A). Results from MTT assay showed that viability of H460 cells co-treated with morusin and bafilomycin A1 was significantly decreased than that of cells treated with morusin alone (Fig. 2B). Similar results were obtained when the percentage of survived cells was measured by trypan blue exclusion assay (Fig. 2C). In addition, co-treatment of morusin and bafilomycin A1 synergistically increased the percentage of sub-G1 phase of H460 cells, indicating that suppression of autophagy obviously could stimulate morusin-induced apoptosis (Fig. 2D). Consistent with the above results, the rate of annexin V-positive H460 cells co-treated with morusin and bafilomycin A1 was markedly increased up to 72.28%, whereas that treated with morusin alone was only 45.7% (Fig. 2E). Taken together, our findings demonstrate that morusin-induced autophagy can protect NSCLC cells from apoptosis.
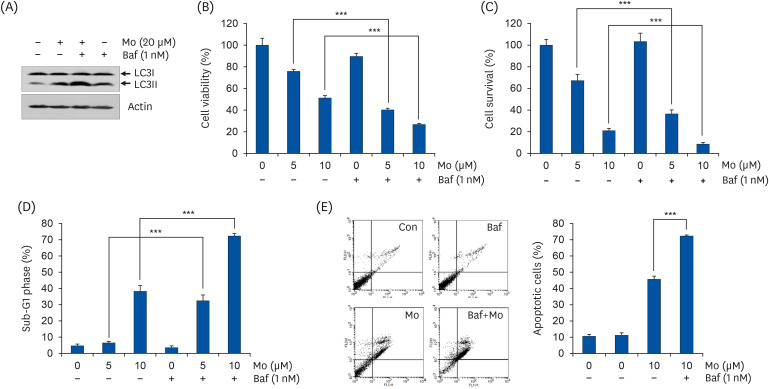 | Fig. 2Inhibition of autophagy enhances morusin-induced apoptosis in non-small cell lung cancer cells. H460 cells were treated with indicated concentration of morusin for 12 h (A) or 72 h (B-E) w/ or w/o bafilomycin A1 (1 nM), an autophagy inhibitor. (A) The expression of LC3II was detected by western blot analysis. Actin was used as an internal control. (B) Cell viability was measured by MTT assay. (C) Percentage of survived cells was evaluated by trypan blue exclusion assay. (D) Cell cycle analysis was performed by flow cytometry to measure the percentage of sub-G1 phase cells (apoptotic cells). (E) Annexin V/PI double staining assay was conducted to detect apoptosis. Annexin V(+) cells were determined as apoptotic cells.
Mo, morusin; Baf, bafilomycin A1.
***P < 0.001 vs. respective controls.
|
AMPK/mTOR pathway mediates morusin-induced autophagy in NSCLC cells
It is well recognized that the AMPK/mTOR signaling axis plays a pivotal role in the process of autophagy. Activation of AMPK suppresses the activity of mTOR, leading to the induction of autophagy and suppression of cell growth [1415]. To determine whether AMPK/mTOR pathway was involved in morusin-induced autophagy, we examined phosphorylation levels of AMPK, mTOR, and their downstream molecules. As shown in Fig. 3, phosphorylation of AMPK was obviously increased by morusin in a time-dependent manner in both H460 and H1299 cells. The expression of p-ACC, a downstream molecule of AMPK, was also enhanced until 24 h post treatment with morusin, although it was reduced at 48 h probably due to reduction of total protein. By contrast, phosphorylated/total ratios of mTOR and its downstream molecule p70S6K were significantly decreased time-dependently after morusin treatment in both H460 and H1299 cells (Fig. 3A and B). Collectively, these results demonstrate that morusin-induced autophagy is mediated by activation of AMPK and suppression of mTOR pathway.
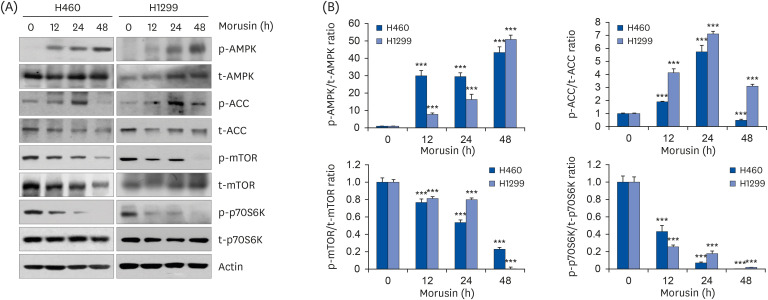 | Fig. 3Role of AMPK/mTOR pathway in morusin-induced autophagy in non-small cell lung cancer cells. Cells were treated with morusin (20 μM) at various time periods. (A) The expression of indicated protein was detected by western blot. Actin was a loading control. (B) The ratio between phosphorylated proteins and total proteins was calculated after densitometric analysis of immunoblots using Image J software.
AMPK, AMP-activated protein kinase; ACC, acetyl-CoA carboxylase; mTOR, mammalian target of rapamycin; p70S6K, phospho-p70S6 kinase.
***P < 0.001 vs. untreated controls.
|
Inhibition of AMPK activity enhances morusin-induced apoptosis in NSCLC cells
Previous studies have reported that AMPK activation triggers not only autophagy, but also apoptosis of cancer cells [26]. Our previous study and current results demonstrate that morusin can induce both apoptosis and autophagy in NSCLC cells [20]. Thus, we next investigated whether morusin-induced AMPK activation was involved in autophagy induction as a survival mechanism or involved in apoptosis induction as a death mechanism in NSCLC cells. As shown in Fig. 4A, phosphorylation levels of both AMPK and ACC were significantly decreased by compound C, an inhibitor of AMPK, indicating that compound C could effectively inhibit the kinase activity of AMPK (Fig. 4A). In addition, our results showed that treatment with compound C enhanced the cytotoxic effect of morusin in H460 cells based on MTT assay (Fig. 4B) and trypan blue exclusion assay (Fig. 4C). Viability of H460 cells co-treated with morusin and compound C was significantly reduced compared to that of cells treated with morusin alone (Fig. 4B and C). In addition, suppression of AMPK by compound C enhanced morusin-induced apoptosis of H460 cells. The proportion of sub-G1 phase was markedly increased up to 23.78% after co-treatment with morusin and compound C. It was only 10.81% in cells treated with morusin alone (Fig. 4D). Taken together, our findings suggest that AMPK activation by morusin can function as a defense mechanism against morusin-induced apoptosis probably by inducing autophagy in NSCLC cells.
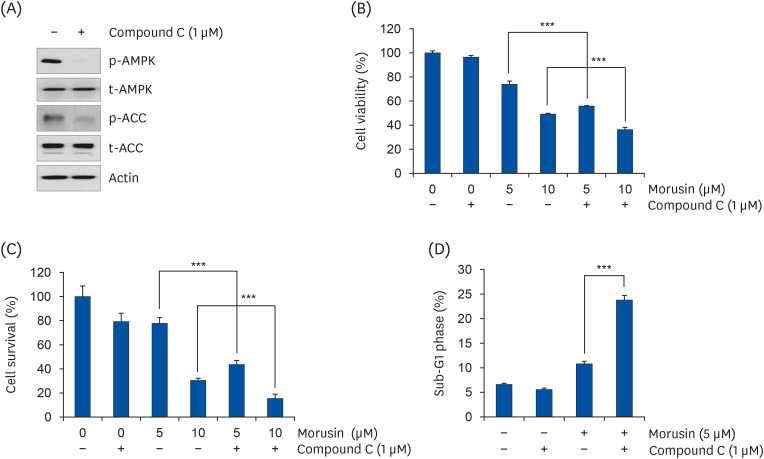 | Fig. 4Inhibition of AMPK enhances morusin-induced apoptosis in non-small cell lung cancer cells. H460 cells were incubated with morusin (5 μM) for 24 h (A) or 72 h (B-D) w/ or w/o compound C (1 μM), an AMPK inhibitor. (A) Expression of indicated protein was detected by western blot analysis. Actin was used as an internal control. (B) Cell viability was examined by MTT assay. (C) The percentage of survived cells was evaluated by trypan blue exclusion assay. (D) The percentage of sub-G1 phase cells (apoptotic cells) was measured by flow cytometry.
AMPK, AMP-activated protein kinase; ACC, acetyl-CoA carboxylase.
***P < 0.001 vs. respective controls.
|
Go to :
DISCUSSION
A growing knowledge about controversial roles of autophagy in cancer progression has driven clinical trials to induce or inhibit autophagy as a probable therapeutic strategy for cancer treatment. In the current study, we investigated the function of morusin-induced autophagy in the survival of NSCLC cells and explored the underlying molecular mechanism. Main findings of this research are as follows. First, we demonstrated that morusin-induced autophagy was a defense mechanism against morusin-induced apoptosis in NSCLC cells. Although Cho et al. [21] have reported that morusin can trigger autophagy in ovarian cancer cells, this is the first study that verifies the role and the underlying mechanism of moruisn-induced autophagy in NSCLC cells. Second, the current study emphasized the role of AMPK in autophagy induction and survival of NSCLC cells under cytotoxic stress. Activation of AMPK and subsequent deactivation of mTOR mediated morusin-induced autophagy, finally protecting NSCLC cells from apoptosis.
Although autophagy can act as a tumor suppressor by inducing autophagic cell death, a type II programmed cell death, in cancer cells [7], our results clearly demonstrate that morusin-induced autophagy can function as a survival mechanism in NSCLC cells, suggesting that the efficacy of morusin might be enhanced by combination with other autophagy inhibitors. Currently, chloroquine (CQ) and hydroxychloroquine (HCQ) are clinically available autophagy inhibitors. They can deacidify the lysosome and block fusion between lysosomes and autophagosomes [27]. A variety of clinical trials have reported that CQ or HCQ in conjunction with other chemotherapies can improve clinical outcomes, prolonging overall survival and increasing progression-free survival [10].
AMPK plays a critical role in the process of autophagy. It has been recognized as a novel target for cancer treatment. Numerous studies have reported a close relationship between AMPK and tumor progression [26]. Basically, AMPK attenuates cell proliferation by blocking the activity of mTORC1 [14]. As 30%–50% percent of NSCLC cases have LKB1 mutation [2829], it has been hypothesized that inability to activate AMPK can result in unsuppressed cell proliferation in NSCLC [1430]. William et al. [31] have reported that AMPK activation in NSCLC is correlated with a better prognosis and an increase in overall survival. In addition, various AMPK agonists such as metformin and AICAR exhibit strong anti-tumor activities [32]. Conversely, induction of autophagy by AMPK activation could result in active growth of tumors by maintaining energy production, which offers a potential drawback of AMPK activation in cancer therapy [33]. Our previous study [20] and current results showed that morusin could trigger both apoptosis and cytoprotective autophagy in NSCLC cells. Thus, we investigated whether morusin-induced AMPK activation could lead to cell survival or apoptotic cell death. Our results clearly showed that inhibition of AMPK enhanced the cytotoxic effect of morusin in NSCLC cells, suggesting that AMPK functioned as a defense mechanism against morusin-induced apoptosis probably by inducing autophagy in NSCLC cells. Regarding mediators of morusin-triggered apoptosis in cancer cells, several candidates including EGFR, mitogen-activated protein kinase, nuclear factor kappa B, and signal transducer and activator of transcription 3 have been suggested instead of AMPK activation [1920].
Interestingly, our previous study has demonstrated that methylene chloride extract of Morus alba L. (MEMA) can trigger autophagy in NSCLC cells as a survival mechanism through ROS generation [22]. We have also shown that morusin is present in MEMA as a marker component of Morus alba L. [34], suggesting that morusin can be an active compound of MEMA that induces autophagy in NSCLC cells. Whether ROS is involved in morusin-induced autophagy should be further determined. Given that the accumulation of ROS can activate AMPK to exert discrepant impacts on the survival of cancer cells [35], it is possible that ROS is the mediator of morusin-induced autophagy in NSCLC cells. However, our preliminary data showed that pretreatment with N-acetylcysteine in NSCLC cells did not enhance morusin-induced apoptosis (data not shown). This observance suggests that the concrete mechanism underlying MEMA-induced autophagy can be different from that of morusin-induced autophagy.
Whether morusin is effective in chemo-resistant NSCLC cells is an important question. In this regard, our previous study has demonstrated that morusin can induce apoptosis in EGFR tyrosine kinase inhibitor-resistant NSCLC cells and treatment-naive NSCLC cells [20]. Although the current study does not focus on the chemoresistant-overcoming activity of morusin, autophagy has emerged as a potential mechanism involved in drug resistance in NSCLC [3637]. Therefore, combined treatment with morusin and autophagy inhibitor might be effective for chemoresistant NSCLC cells. This hypothesis should be verified by a future study. A lack of in vivo experiments is a drawback of this study. Reproducibility in an in vivo model can be a strong evidence to support our conclusion. Therefore, it is our next goal to verify the autophagy-inducing activity of morusin in vivo and to investigate whether autophagy inhibition can enhance the efficacy of morusin in a mouse model.
In conclusion, the current study demonstrated that morusin could induce autophagy in NSCLC cells through AMPK activation as a defense mechanism against morusin-induced apoptosis. This study provides some insight into controversial roles of autophagy and AMPK in cancer progression. Although further studies are needed to evaluate anticancer effects of morusin, our results suggest that combination treatment of morusin with autophagy inhibitor or AMPK inhibitor may enhance the clinical efficacy of morusin in NSCLC.
Go to :
 XML Download
XML Download