

 PDF
PDF ePub
ePub Citation
Citation Print
Print



ABBREVIATIONS
ALL
acute lymphocytic leukemia
CIMP
CpG island methylator phenotype
CRC
colorectal cancer
DCC
deleted in colorectal carcinoma
DDATHF
5,10-dideazatetrahydrofolate
FA
folic acid
FPGS
folylpolyglutamate synthase
5FdUMP
5-fluoro-2-deoxyuridine-5-monophosphate
5FU
5-fluorouracil
GGH
γ-glutamyl hydrolase
HDAC
histone deacetylase
LV
leucovorin
MTX
methotrexate
NHANES
National Health and Nutrition Examination Survey
RA
rheumatoid arthritis
RFC
reduced folate carrier
siRNA
small-interfering RNA
SNPs
single-nucleotide polymorphisms
THF
tetrahydrofolate
TS
thymidylate synthase
UL
tolerable upper intake level
INTRODUCTION
Folate is a water-soluble B vitamin that acts as an important mediator of one-carbon transfer and plays a crucial role in human health and disease [1]. Naturally occurring folates are present in green leafy vegetables, asparagus, broccoli, legumes, whole grain, and organ meats and are extremely unstable and easily oxidized in low pH conditions. In contrast, folic acid (FA) is the most stable and a fully oxidized monoglutamyl synthetic form of folate that is commonly used in commercial supplements and fortified food products [1]. Ever since the mandate on FA fortification of white wheat flour, cereal, and enriched pastas was passed in 1998, grain products have been used as a major source of folate in the United States and Canada [2]. Folate deficiency is reportedly associated with the development of neural tube defects and congenital disorders, anemia, atherosclerosis, adverse pregnancy outcomes, neuropsychiatric disorders, and cognitive impairments [3]. Accumulating epidemiological evidence suggests an inverse relationship between dietary folate intake or blood folate levels and the risk of multiple malignancies, including lung, oropharyngeal, esophageal, stomach, colorectal, prostate, and breast cancer [34567]. However, clinical and preclinical studies suggest that excess FA consumption is associated with an increased risk of progression of established precancerous lesions, indicating that folate may play a dual modulatory role in cancer development and progression [568910].
A study using the National Health and Nutrition Examination Survey (NHANES) 2003-2006 data (n = 11,462) reported that 34.5% of the population in the United States used dietary supplements containing FA. The use of dietary supplements containing FA was highest in individuals aged 51-71 years, with 5% of the individuals consuming FA above the tolerable upper intake level (UL) from dietary supplements alone [11]. Another study that used data from the Canadian Community Health Survey 2.2 (n = 34,381) revealed that 25% of the study population consumed supplements containing FA, and 9-14% of individuals aged 14 years or above consuming vitamin/mineral supplements had a consumption above the permissible UL for FA [12]. Furthermore, use of supplements by cancer survivors is relatively common. The previous systematic review reported that 64–81% of cancer survivors used a vitamin or mineral supplement and 14–32% of survivors started taking supplements after diagnosis [13]. Several studies on the use of FA supplements have been conducted on colorectal cancer (CRC) survivors. In CRC survivors, the consumption of FA-containing supplements increased from 35.4% to 55.1% after diagnosis, while consumption of FA or FA-containing supplements was higher among female survivors [141516]. However, a growing body of evidence suggests that folate supplementation enhances efficacy of anticancer drugs, such as 5-fluorouracil (5FU), and helps in management of treatment toxicity due to antifolates, whereas increased folate levels may interfere with the efficacy of chemotherapeutic agents and induce drug resistance [17181920]. This review discusses the effects of enzymes associated with maintenance and distribution of intracellular folate on antifolates- and 5FU-based cancer chemotherapy.
Intracellular folate homeostasis
Monoglutamates are the only form of folate present in circulation, while intracellular folate molecules primarily exist as polyglutamates after cellular uptake by transporters such as reduced folate carrier (RFC), folate receptor, and proton-coupled folate transporter [1]. Upon its entry into the cell, the folate molecules are linked to glutamate residues by the enzyme folylpolyglutamate synthase (FPGS) through a process known as polyglutamylation; this facilitates the retention of folate in the cell and helps maintain a steady supply of utilizable folate derivatives for folate-dependent enzyme reactions. γ-glutamyl hydrolase (GGH) is an enzyme that catalyzes the hydrolysis of polyglutamylated folate into monoglutamylated folate that is subsequently exported from the cell. Both FPGS and GGH are critical enzymes that function synergistically to maintain intracellular folate concentration and distribution [121].
Folate and cancer treatment
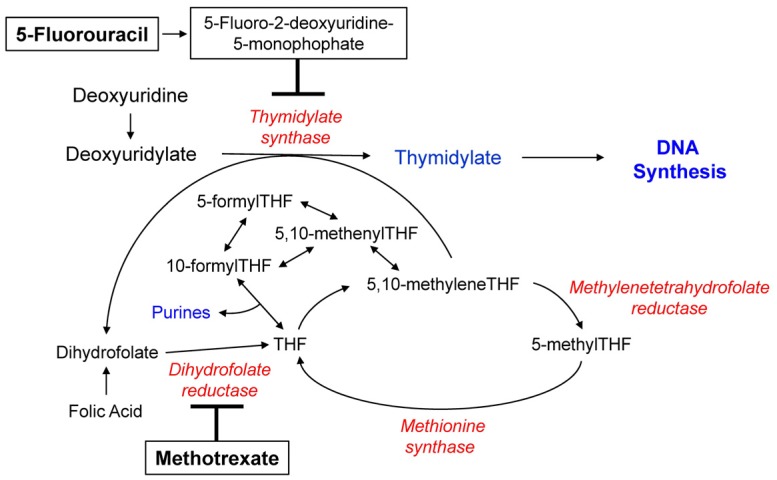
Folate is an essential cofactor for DNA synthesis; therefore, folate depletion and disrupted folate metabolism can reduce the efficacy of DNA synthesis, resulting in inhibition of tumor growth in neoplastic cells; this theory forms the basis for cancer chemotherapy using antifolates and 5FU [22]. Antifolates, such as methotrexate (MTX), used for the treatment of human malignancies and rheumatoid arthritis (RA) are structurally similar to folate and act by binding to or inhibiting folate-dependent enzymes [23]. MTX typically acts by inhibiting dihydrofolate reductase and reducing the intracellular folate concentration necessary for thymidylate and purine synthesis, eventually preventing DNA synthesis (Fig. 1). Additionally, MTX polyglutamates can directly inhibit thymidylate synthase (TS) and enzymes involved in de novo purine biosynthesis [2425]. Similar to folates, polyglutamylated antifolates exhibit better retention in cells, thereby increasing the cytotoxicity of antifolates by increasing the duration of exposure [2122]. Moreover, polyglutamylated antifolates inhibit their target folate-dependent enzymes involved in thymidylate and purine biosynthesis more efficiently as the polyglutamylated forms exhibit higher affinity for these enzymes compared to the monoglutamated forms [2122].
5FU, a prototype of pyrimidine antagonists, is widely used in the treatment of colon and breast cancer [26]. One of the cytotoxic mechanisms adopted by 5FU is the formation of a ternary complex involving 5-fluoro-2-deoxyuridine-5-monophosphate (5FdUMP, a metabolite of 5FU), TS, and 5,10-methylenetetrahydrofolate (5,10-methyleneTHF) [27]. This ternary complex suppresses TS activity with consequent depletion of intracellular thymidylate reserves, resulting in inhibition of DNA synthesis (Fig. 1) [27]. Leucovorin (LV; 5-formylTHF), a precursor of 5,10-methyleneTHF, potentiates 5FU cytotoxicity by stabilizing the inhibitory ternary complex (Fig. 1) [27]. 5,10-methyleneTHF with longer chain polyglutamates exhibit greater potential for the formation and stabilization of the 5,10-methyleneTHF-TS-5FdUMP ternary complex than those with shorter chain polyglutamates, suggesting that 5,10-methyleneTHF polyglutamylation may influence 5FU efficacy [28].
A growing body of evidence suggests that FPGS and GGH affect chemosensitivity of cancer cells to antifolates and 5FU by altering duration of intracellular retention of antifolates and 5,10-methyleneTHF (a specific target folate cofactor for 5FU), respectively [2930]. In addition to their implication in polyglutamylation, FPGS and GGH alter the intracellular folate status, which is a critical determinant of chemosensitivity of cancer cells to chemotherapeutic agents designed to interrupt intracellular folate metabolism and DNA synthesis [293031]. Therefore, FPGS and GGH play a substantial role in the maintenance of intracellular homeostasis of folates and antifolates for optimal folate-dependent one-carbon transfer reactions and antifolate-induced cytotoxic effects.
Folate mediates the transfer of one-carbon units in DNA methylation as well, which might influence chemotherapeutic effects by altering expression of genes involved in drug response [132]. Given that polyglutamylated folates act as better substrates for enzymes involved in DNA methylation, such as methyleneTHF reductase and methionine synthase, polyglutamylation plays a pivotal role in DNA methylation [2133] (Fig. 1). Therefore, FPGS- and GGH-mediated polyglutamylation-induced changes in DNA methylation might affect chemosensitivity to chemotherapeutic agents.
Effects of folylpolyglutamate synthase on cancer chemotherapy
Several studies report the effects of differential FPGS activity and FPGS modulation on drug resistance of and chemosensitivity to antifolates such as MTX and 5FU. Considering that FPGS has a lower affinity for MTX compared to that for folates (dihydrofolate > THF > 5-methylTHF > MTX), the formation of MTX polyglutamates is slower than that of folate polyglutamates. Therefore, reduced FPGS activity would affect folate polyglutamate pools less significantly and critically reduce MTX cytotoxicity [3435]. In general, high FPGS activity or its upregulation seems to enhance chemosensitivity of cancer cells to MTX and 5FU, whereas low FPGS activity or downregulation appears to correspond to resistance to these drugs. FPGS overexpression was associated with enhanced MTX efficacy in AUXB1 variant hamster cells lacking endogenous FPGS activity [36]. Upregulation of FPGS gene expression increased sensitivity to MTX and other antifolates in 9L rat gliosarcoma cells [37]. On the contrary, FPGS inhibition has been suggested as the mechanism underlying resistance to antifolates such as MTX in human and murine leukemia cells [383940414243]. Reduced FPGS activity was associated with resistance to 5FU in CCRF-CEM human acute lymphocytic leukemia (ALL) and HCT8 human colon adenocarcinoma cells [444546]. Sohn et al. [47] reported that FPGS overexpression increases and FPGS inhibition decreases the chemosensitivity of HCT116 human colon adenocarcinoma cells toward 5FU. The same group of authors highlighted that in MDA-MB-435 human breast adenocarcinoma cells, FPGS overexpression increases chemosensitivity to MTX, while FPGS inhibition reduces 5FU-induced chemosensitivity at supraphysiologial folate medium concentrations [29]. In addition, FPGS modulation affects polyglutamylation of antifolates and specific target folate cofactors (for example, 5,10-methyleneTHF for 5FU), as well as of intracellular folate cofactors, which are critical determinants of antifolate and 5FU cytotoxicity [2229474849]. FPGS plays an important role in cancer cell sensitivity to antifolates and 5FU; therefore, FPGS modulation might be a potential therapeutic target for increasing cancer cell sensitivity to these chemotherapeutic agents.
Altered FPGS expression was reported to be associated with CRC patient outcome. Odin et al. [50] reported that the expression of RFC, FPGS, GGH, and TS increased in CRC tumor biopsies compared to that in the adjacent non-neoplastic mucosa. Moreover, they revealed the association between low expression of RFC and FPGS and the absence of the mRNA splice variant of the putative tumor suppressor gene deleted in colorectal carcinoma (DCC) in normal-appearing mucosa of CRC patients [51]. Although several single-nucleotide polymorphisms (SNPs) have been reported in the FPGS gene, there are a limited number of studies on the functionality or frequencies of these SNPs [5253]. The mutant Cys346Phe FPGS reportedly interferes with L-glutamate or ATP binding, resulting in disruption of FPGS activity [41]. The CC genotype of FPGS rs1544105C>T was associated with poor response to MTX in patients with RA and pediatric B-cell precursor ALL [5455]. Furthermore, in lymphoid cells, FPGS expression was epigenetically regulated by chromatin remodeling through interactions between NFY, Sp1, and histone deacetylase (HDAC) factors binding to the FPGS promoter region, resulting in low intracellular accumulation of long-chain MTX polyglutamates [56]. Conversely, HDAC inhibitors increased FPGS expression and long-chain MTX polyglutamate accumulation in childhood ALL cells [57]. In addition, FPGS modulation altered DNA methylation and expression of genes associated with folate biosynthesis and one-carbon metabolism along with increased 5FU efficacy in response to FPGS overexpression [2958]. This result indicates that FPGS-mediated polyglutamylation-induced changes in DNA methylation might influence chemosensitivity to chemotherapeutic agents as well.
Effects of γ-glutamyl hydrolase on cancer chemotherapy
As mentioned previously, both FPGS and GGH are involved in the maintenance of intracellular folate and antifolate homeostasis. GGH was reported to have a higher affinity for longer chain polyglutamates (for example, Glu4
versus Glu1 derivatives, Km 17- and 15-fold lower for folate and MTX, respectively) in HT-1080 human sarcoma cells [59]. The ratio of GGH/FPGS serves as a reliable predictor of the relative concentrations of long-chain MTX polyglutamates in patients with acute leukemia, whereas the ratio of FPGS/GGH was associated with MTX-Glu4-6 accumulation and MTX resistance in childhood leukemia patients [6061]. A growing body of evidence suggests the association between lower FPGS and/or higher GGH activity and reduced antifolate polyglutamylation, which is related to drug resistance [6263646566]. Compared to parental H35 cells, an increase of approximately 7-fold in GGH activity and no change in FPGS activity was observed in H35D rat hepatoma cells resistant to the antifolate 5,10-dideazatetrahydrofolate (DDATHF) [64]. In this study, two H35 cell lines had nearly equal intracellular folate levels; however, the polyglutamate chains in H35D cells were predominantly composed of Glu3 and Glu4, while those in H35 cells were primarily composed of Glu5 and Glu6 [64]. Moreover, folate and antifolate accumulation was reduced in H35D cells along with an increase in GGH activity [66]. Similarly, the resistance in MTX-resistant HT-1080 and DDATHF-resistant CCRF-CEM human leukemia cells was attributed to increased GGH activity [6263]. In addition, Kim et al. [30] demonstrated that GGH overexpression reduced the chemosensitivity of HCT116 and MDA-MB-435 cells to MTX and 5FU, which was affected by exogenous folate levels.
Conversely, there are several studies that highlight the association between a reduced GGH activity and an increased accumulation of long-chain MTX polyglutamates. A previous study reported that cellular and secreted GGH levels were reduced by 60% in the presence of insulin in H35 cells [67]. Given that the presence of insulin increased MTX glutamylation by 3-fold in intact H35 cells [6869], O'Connor et al. [67] suggested that insulin-induced polyglutamylation enhancement might be related to the reduction in GGH levels. This explanation was further validated by a study that indicated an inverse relation between insulin-induced reduction in GGH activity and the changes in intracellular synthesis and accumulation of MTX polyglutamate [70]. An inverse association was observed between GGH activity and the accumulation of total and long-chain (Glu4-7) MTX polyglutamates in the blast cells of ALL patients administered a high dose of MTX.
Furthermore, reduction in GGH expression in DLD-1 human colon cancer cells induced by small-interfering RNA (siRNA) increased sensitivity to 5FU+LV or only to 5FU, presumably due to improved retention of 5,10-methyleneTHF [31]. In this study, siRNA- induced FPGS downregulation reduced the basal levels of reduced folate, the accumulation and retention of folate in LV-treated DLD-1 cells, and the sensitivity to 5FU+LV. These results suggest that tumors expressing high levels of FPGS and low levels of GGH are likely to be most sensitive to 5FU-induced chemosensitivity in the presence of LV [31]. Another study reported that siRNA-induced GGH inhibition significantly enhanced chemosensitivity of HCT116 and MDA-MB-435 cells to 5FU and the chemotherapeutic effect was augmented by the increase in exogenous folate levels [30]. In addition, the CpG island methylator phenotype (CIMP)+, accounting for approximately 17% of CRC cases [71], is associated with low GGH expression and high 5,10-methyleneTHF levels compared to CIMP- tumors, which correspond to positive response of CIMP+ tumors to 5FU [7273]. However, FPGS levels did not differ significantly between CIMP+ and CIMP- tumors [73], suggesting that FPGS might have a lesser impact than GGH on chemosensitivity of CRC to 5FU.
Several SNPs identified in the GGH gene bases have been shown to compromise the promoter as well as the coding regions of the gene [74]. These promoter polymorphisms were associated with increased GGH expression in HepG2 human hepatoma cells. -401C>T and -124T>G showed enhanced GGH expression in MCF7 human breast cancer cells, suggesting that polymorphisms in the GGH gene promoter may increase GGH expression [74]. Moreover, the GGH-401 T allele frequency varied among ethnic or populations groups in healthy adults-22-36% in Asian, 31% in Caucasian, and 14% in African population groups [75]. In RA patients, patients with the TT genotype of -401C>T were associated with higher GGH activity compared to patients with CC or CT genotypes, indicating that the TT genotype might be related to poor response to antifolates [76]. However, in Korean patients with cervical cancer who underwent radical hysterectomy, a poor response to platinum-based neoadjuvant chemotherapy was associated with the CC genotype of -401C>T, which might be attributed to the counteractive effects of CC genotypes of XRCC1 A194T and GGH -401C>T in cervical cancer [77]. Furthermore, there was an interethnic difference in the GGH 452 T allelic frequency- 14% in Caucasian, 9-10% in Asian, and 8% in African population groups [75]. GGH +452C>T was associated with low GGH activity and high MTX polyglutamate accumulation in ALL patients [78]. In addition to SNPs, epigenetic regulation can modulate GGH activity and MTX polyglutamate accumulation in human leukemia cells. Methylation of two CpG islands (CpG1 and CpG2) in the GGH promoter was associated with reduced GGH mRNA expression/activity and higher accumulation of MTX polyglutamates in human acute leukemia cells, which would suitably explain the better response to antifolates [7980]. Previous studies that adopted epigenomic and genomic approaches suggested that GGH modulation-induced alterations in concentrations of total intracellular and long-chain polyglutamylated folate affected global and gene-specific DNA methylation and gene expression that was partially associated with 5FU efficacy [3081]. When considered collectively, it appears that identified and characterized GGH SNPs and/or GGH-induced epigenetic changes contribute to functional and pharmacological consequences in cancer cells.
CONCLUSIONS
Notwithstanding the complexity of FPGS- and GGH-induced changes in folate and antifolate accumulation and metabolism, the potential roles played by genetic variants of these enzymes in antifolate- and 5FU-based cancer chemotherapy warrants further investigation. Elucidating the pharmocogenetic ramifications of these enzyme-induced changes will provide a framework for developing rational, effective, safe, and customized chemotherapeutic practices.
 XML Download
XML Download