

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Oxidative stress is involved in the pathogenesis of diseases such as cardiovascular disease, chronic kidney disease, neurodegenerative diseases (NDs), macular degeneration, and cancer [1]. Reactive oxygen species (ROS) generated during normal metabolic processes of cells are removed by intracellular antioxidant enzymes, but imbalances between the production and elimination of ROS that result from abnormalities of the antioxidant homeostasis system induce oxidative stress [234]. Oxidative stress causes peroxidation of lipids in the cell membrane, impairment of intracellular essential components such as proteins and DNA, induction of mitochondrial dysfunctions, and activation of apoptosis-related cell signals, eventually leading to fatal injury and cell death [56].
NDs including Alzheimer's disease (AD) and Parkinson's disease (PD) lead to progressive loss of cognitive and behavioral capacities, resulting in severe disability and death [7]. Although pathological mechanisms involving NDs are complex and have not yet been fully elucidated [6], apoptosis induced by oxidative stress and abnormal protein aggregation are major causes of pathogenesis [789]. Thus, protecting neuronal cells from injury by oxidative stress is considered a promising therapeutic approach for the treatment of NDs in terms delaying progression and improving status of disease [10].
According to epidemiological studies and many in vitro and in vivo intervention studies, dietary polyphenols have many health benefits such as antioxidant, anti-inflammatory, anticancer, anti-obesity, anti-diabetic, and neuroprotective effects [3]. However, because only 5–10% of dietary polyphenols are absorbed in the small intestine, to apply as dietary agents for the prevention or treatment of diseases has the limitation [1112]. Several studies have recently reported that the health benefits of polyphenol-rich foods are mainly due to their gut microbial-derived metabolites, not their polyphenol compounds [1213]. Polyphenols are converted into phenolic metabolites of small molecular weight by the action of gut microbiota in the small intestine; these gut microbial-derived metabolites have high bioavailability and permeability of the blood-brain barrier (BBB) [14].
Ellagitannins (ETs) are hydrolyzed into ellagic acid (EA) in the body after ingestion of polyphenols in pomegranates, walnuts, and berries. ETs and EA have excellent antioxidant and cell-protective abilities but they are also limited in having low bioavailabilities; thus, studies have increasingly investigated gut microbial-derived metabolites such as urolithins (uros) [1516]. Uros have been found in various forms such as Uro-M5, Uro-M6, Uro-M7, Uro-D, Uro-C, Uro-B, Uro-A, and isoUro-A. Among them, Uro-A (UA) is the most common form in humans. It has health benefits such as antioxidant, anti-cancer, anti-inflammation, and anti-obesity effects in vivo and in vitro [1718]. However, there have been no reports on the effects of UA on brain-related diseases, such as NDs caused by oxidative stress.
In this study, we investigated the protective effects of UA against H2O2-induced oxidative stress in human neuroblastoma SK-N-MC cells. We also examined possible mechanisms associated with the action of UA.
MATERIALS AND METHODS
Materials
The SK-N-MC human neuroblastoma cell line was purchased from American Type Culture Collection (Rockville, MD, USA). Eagle's minimum essential medium (EMEM), trypsin-EDTA, antibiotics, Dulbecco's phosphate-buffered saline (PBS), and Hank's balanced salt solution (HBSS) were purchased from WelGENE (Daegu, Republic of Korea). Fetal bovine serum (FBS), urolithin A (UA), and general reagents were purchased from Sigma-Aldrich (St. Louis, MO, USA). Cell counting Kit-8 (CCK-8) assay reagents were purchased from Dojindo Molecular Technologies (Gaithersburg, MD, USA), and the intracellular ROS assay kit was purchased from Cell Biolabs (San Diego, CA, USA). Anti-Bax (#2772), anti-total p38 (#9212), anti-caspase-9 (#9502), anti-caspase-3 (#9665), anti-PARP (poly (ADP-ribose) polymerase, #9532), anti-mouse IgG horseradish peroxidase [HRP]-conjugated antibody (#7076), anti-rabbit IgG HRP-conjugated antibody (#7074), and p38 mitogen-activated protein kinase (MAPK) inhibitor SB203580 (#5633) were purchased from Cell Signaling Technology (Danvers, MA, USA).
Cell culture and treatment
SK-N-MC cells were grown in EMEM supplemented with 10% FBS and 1% antibiotics, and maintained in a humidified incubator at 37℃ in an atmosphere of 5% CO2 and 95% air. The cell culture medium was changed every 2 days. When the cells were approximately 90% confluent, they were subcultured in plates at an appropriate density according to each experimental scale. The cells were pretreated with various concentrations (1.25, 2.5, and 5 µM) of UA for 6 h and then exposed to 300 µM H2O2 for 18 h.
Measurement of cell viability
The cell viability was evaluated using a CCK-8 assay [19]. CCK-8 is reduced by dehydrogenases in cells to give a yellow-colored product (formazan). The amount of the formazan dye generated by the activity of dehydrogenases in cells is directly proportional to the number of living cells. This characteristic can be used for cell viability analysis. The cells were seeded at 5 × 104 cells/100 µL in a 96-well plate and cultured for 24 h to confirm cell viability. After the cultured cells had been treated with various concentrations of UA and H2O2 for various times, they were incubated with the CCK-8 solution for 2 h. Then absorbance at 450 nm was measured using a microplate reader (Sunrise; Tecan, Grödig, Austria).
Measurement of intracellular ROS production
To measure the amount of ROS produced in cells, used the cell-permeable fluorogenic probe 2′, 7′-Dichlorodihydrofluorescin diacetate (DCFH-DA). In brief, DCFH-DA is diffused into cells and is deacetylated by cellular esterases to non fluorescent DCFH, which is rapidly oxidized to highly fluorescent DCF by ROS [20]. The cells were seeded at 8 × 104 cells/100 µL in a 96-well black plate. After 24 h, the cells were treated with 1.25, 2.5, and 5 µM UA and cultured for 6 h. The cell culture medium was removed and the cells were washed twice with HBSS, treated with 10 µM DCFH-DA solution, and incubated for 45 min. The DCFH-DA solution was removed and the cells were washed twice with HBSS and then treated with 300 µM H2O2. Intracellular fluorescence intensities were measured at 485 nm and 530 nm using a fluorescence microplate reader (Infinite M200; Tecan).
Hoechst 33342 staining
Hoechst 33342 (Invitrogen, Carlsbad, CA, USA) staining was used to observe changes in the nuclei of apoptotic cells [21]. The cells were seeded at 4 × 105 cells/mL in a six-well plate, cultured for 2 days, then treated with various UA concentrations for 6 h. The medium containing UA was removed and the cells were cultured for 18 h in the presence of 300 µM H2O2. Then the cells were washed with PBS and fixed with 4% paraformaldehyde (Sigma-Aldrich) for 10 min at room temperature. After washing with PBS, Hoechst 33342 dye was used at a concentration of 5 µg/mL for 10 min to stain the cells. After staining and washing with PBS, the cells were observed using fluorescence microscopy (IX81; Olympus, Tokyo, Japan).
Western blotting analyses
After removing the culture medium from the treated cells, the cells were washed with PBS, and the proteins were extracted using an EzRIPA lysis kit (ATTO, Tokyo, Japan). Protein concentrations were quantified using the BCA method (Thermo Scientific, Rockford, IL, USA) and then the cells were mixed with 5× SDS sample buffer, followed by loading of 15 µg protein per sample well. The proteins were separated by electrophoresis on a 10–15% SDS-PAGE gel and transferred to a PVDF membrane (Merck Millipore, Darmstadt, Germany). After transfer, the membrane was blocked for 1 h with 5% skim milk (Sigma-Aldrich) in TBS-T buffer. Then the membranes were incubated with primary antibodies diluted at a ratio of 1:1,000, overnight at 4℃. After washing three times, HRP-conjugated secondary antibodies diluted 1:2,000 were added and allowed to react at room temperature for 1 h. After washing three times with TBS-T, protein bands were visualized after treatment with ECL-Western Blotting Substrate (Thermo Scientific).
Statistical analyses
The results are expressed as means ± SD. Statistical analyses included t-tests and one-way analysis of variance (ANOVA) using SPSS statistical software for Windows, version 20.0 (SPSS, Chicago, IL, USA). A one-way ANOVA followed by Duncan's multiple range test was used to determine differences among the treatment groups. The test results were considered statistically significant at P < 0.05.
RESULTS
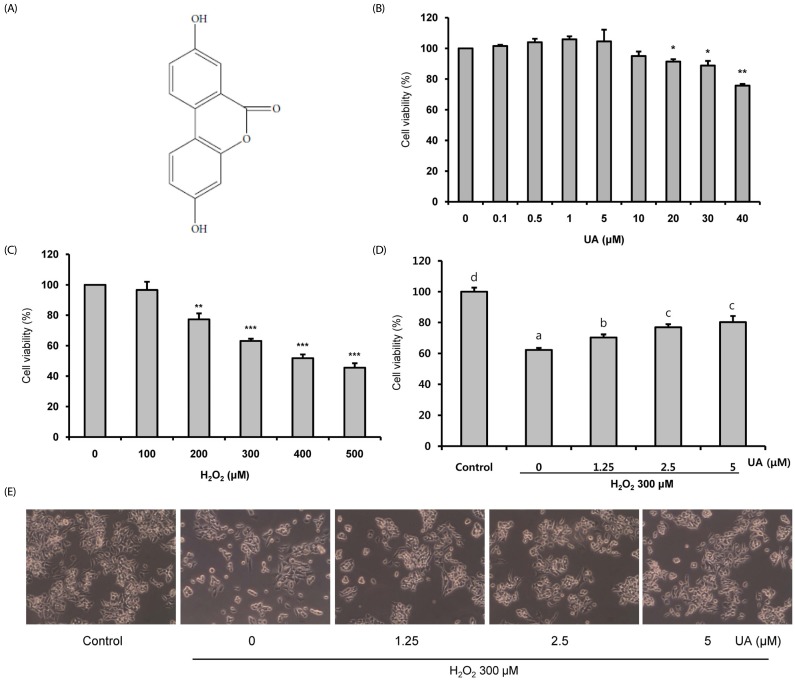
Effect of UA on cell viability
Human neuroblastoma SK-N-MC cells have been widely used as an in vitro model to study the pathogenesis of NDs such as AD because of their high stability and homogeneity [2223]. The cell viabilities were determined using the CCK-8 assay. Cell viability was not significantly affected at UA concentrations up to 10 µM (Fig. 1B). We subsequently chose a UA concentration range of 0.1-10 µM that did not induce cytotoxicity in SK-N-MC cells. H2O2 significantly decreased cell viability in a dosedependent manner; 300 µM indicated 63.1 ± 1.5% cell viability (Fig. 1C). To assess the neuroprotective effects of UA against H2O2, the cells were pretreated with UA for 6 h and then treated with 300 µM H2O2 for 18 h (Fig. 1D). Pretreatment with UA significantly increased cell viability compared to H2O2 alone (62.3 ± 1.3%). Pretreatment with different concentrations of UA (1.25, 2.5, and 5 µM) increased the cell viability to 70.2 ± 2.0, 76.9 ± 2.0, and 80.2 ± 4.0%, respectively. These results were also confirmed by observing cell morphologies (Fig. 1E).
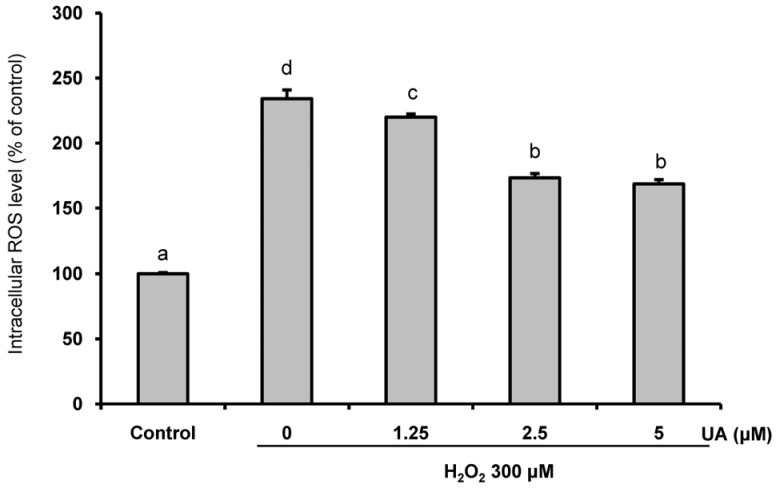
Effects of UA on intracellular ROS production
To confirm that UA inhibited ROS production induced by H2O2, ROS levels in the cells were detected using DCFH-DA. Intracellular ROS production was increased by 2.34 ± 6.69-fold in the group treated with 300 µM H2O2, compared to the controls. However, pretreatment with UA significantly diminished the increase in intracellular ROS production (Fig. 2).
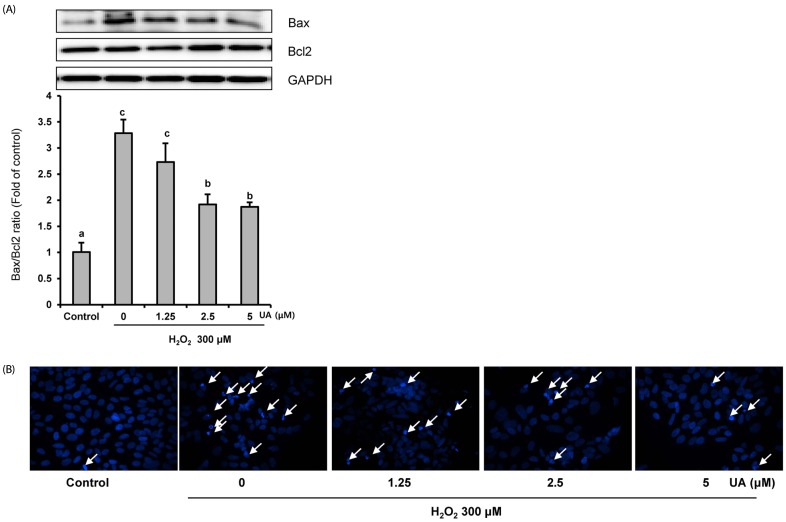
UA inhibits H2O2-induced apoptosis
To demonstrate the anti-apoptotic effects of UA, we confirmed the protein expressions of Bcl2 and Bax via Western blotting analyses. In the 300 µM H2O2 treatment group, the Bax/Bcl2 ratio increased approximately three-fold compared to the control group. However, UA pretreatment resulted in a significant decrease in the Bax/Bcl2 ratio, particularly at UA concentrations of 2.5 and 5 µM, compared to the H2O2-treated group (Fig. 3A).
In addition, Hoechst 33342 staining showed DNA condensation and nuclear fragmentation after H2O2 treatment. However, these apoptotic characteristics were inhibited by pretreatment with UA (Fig. 3B). Taken together, the results imply that UA pretreatment inhibited H2O2-induced apoptosis.
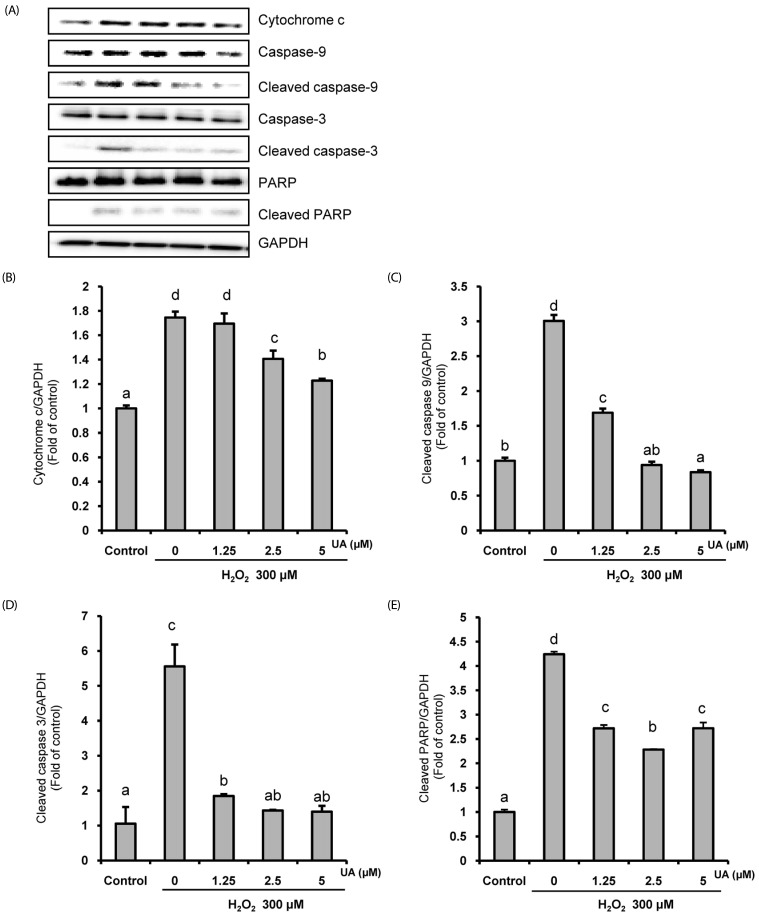
UA prevents apoptotic cell death by suppressing the expression of mitochondrial-related apoptosis proteins
Next, we investigated the protein expression of the mitochondrialrelated apoptosis pathway in H2O2-induced SK-N-MC cells in the presence or absence of UA. As shown in Fig. 4, H2O2 increased the expressions of cytochrome c, cleaved caspase-9, cleaved caspase-3, and cleaved PARP. However, in the UA pretreatment group, the expressions of these mitochondrial-related apoptosis proteins were suppressed, demonstrating that UA attenuates apoptotic cell death by its anti-apoptotic properties against H2O2-induced apoptosis in SK-N-MC cells.
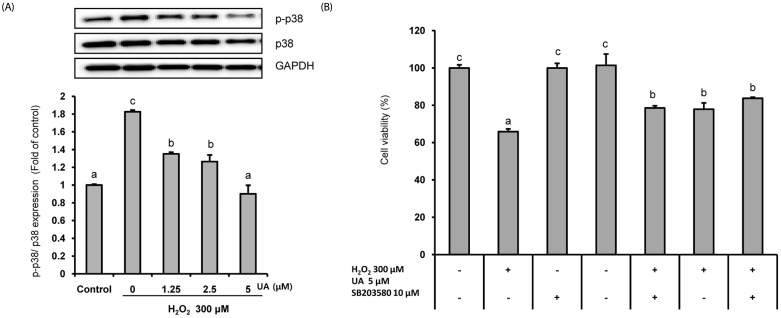
UA has protective effects by modulating the p38 MAPK pathway.
The mitogen-activated protein kinase (MAPK) signal pathway, including c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and p38 was activated by ROS. We investigated whether UA regulated the MAPK signal pathway in SK-N-MC cells. As shown in Fig. 5, pretreatment with UA significantly reduced the expression of p-p38 induced by H2O2. However, the effects on the expressions of p-JNK and p-ERK were not significant (data not shown).
To further confirm the action of UA on the p-p38 MAPK pathway, changes in cell viability were measured by treating cells with a p38 MAPK inhibitor (SD203580) (Fig. 5B). The cell viability decreased to 65.8 ± 1.5% when treated with H2O2, but was significantly increased by treatment with p38 MAPK inhibitor and UA (77.9 ± 3.6% and 78.6 ± 1.2%, respectively). In addition, the cell viability was further increased to 83.7 ± 0.6% in the group pretreated with p38 MAPK inhibitor and UA. Together, the results indicate that UA treatment suppresses H2O2-induced neuronal cell death by modulating the p38 MAPK pathway.
DISCUSSION
Polyphenols are converted into small molecular metabolites by gut microbiota. The converted metabolites have the advantages of excellent body utilization and high BBB permeability, compared to polyphenols [1324]. Therefore, metabolites derived from gut microbes are expected to be effective protective agents for the prevention and treatment of brain-related diseases. In this study, we hypothesized that UA, a representative polyphenol metabolite, may have neuroprotective effects by inhibiting cell death induced by oxidative stress.
ETs, which are abundant in pomegranates, berries, and nuts, are hydrolyzed into EA in the body after ingestion as a kind of water-soluble tannin, and hydrolyzed EA is metabolized by Uros [172425]. ETs and EA have anti-inflammatory, antioxidant, and anti-cancer effects [2627], but dietary EA has low bioavailability, due to its low solubility in the stomach and limited absorption in the intestine [14282930]. González-Sarrías et al. [31] reported that EA concentrations in the plasma or tissues do not exceed 100 nM, even after oral ingestion of pure EA at high concentrations. However, when EA is converted into UA, the uptake in the intestinal villi increases and is maintained at a high level in the plasma, allowing circulation to the target tissue through the blood [112832]. Cerda et al. [33] detected UA levels in plasma of up to 14–40 µM after administering pomegranate juice to healthy people, and there was no apparent toxicity. Vicinanza et al. [34] also reported high concentrations of UA in the target tissues of animal models after oral administration. Therefore, after intestinal absorption, UA has high bioavailability because can reach concentrations in the bloodstream that can exert effects in vivo. Moreover, UAs circulating through the blood can penetrate the BBB. The BBB is a interface that limits and regulates molecular exchanges between the blood and the neuronal tissue or its fluid spaces, having a crucial role in providing nutrients and controlling the access of compounds to the brain [3536]. Although high-molecular-weight polyphenols such as ETs is not able to cross the BBB, UA conversed into low-molecular-weight metabolites has high permeability on the BBB [2837]. UA is the major form found in humans [1732]. It has antioxidant [38], anti-inflammatory [18], and anti-cancer [13293032] effects, attenuates endothelial dysfunction [39], and inhibits fat accumulation [17]. However, few studies on the action of UA in relation to the pathogenesis of NDs due to oxidative stress have been conducted. We expected that UA would has potential a therapeutic effect on the NDs by reducing loss of brain tissue through protection of neuronal cells. Therefore, we investigated the neuroprotective effects of UA on oxidative stress.
UA pretreatment increased the cell viability reduced by H2O2 in SK-N-MC cells (Fig. 1). It also decreased the increased intracellular ROS production (Fig. 2). According to many previous in vitro studies, hydrogen peroxide (H2O2) had toxic effects on various cells, including neurons, by causing oxidative damage to nucleic acids, proteins, and cell membrane lipids [71040]. In our study, UA protected cells against H2O2 toxicity and inhibited the induction of oxidative damage by ROS. UA exhibits antioxidative effects through removal of free radicals [41] and inhibition of pro-oxidative enzymes such as hemeperoxidase [38]. Therefore, we conclude that the antioxidant activity of UA protected SK-N-MC neuronal cells from oxidative damage by H2O2. Tang et al. [18] also reported that when hypoxia/reoxygenation (H/R) injury was induced in primary cultured neonatal rat cardiomocytes, decreased cell viability and increased accumulation of intracellular ROS were attenuated by pretreatment with 10 µM UA. Although there have been several reports that high concentrations (> 50 µM) of UA have antiproliferative effects in various cancer cell lines [1317], a study using SH-SY5Y human neuroblastoma cells reported that pretreatment with 10 µM UA inhibited the reduction of cell viability by H2O2 [11]. Moreover, our results demonstrate that low concentrations (≤ 5 µM) of UA may alleviate inhibition of cell growth by oxidative stress in SK-N-MC human neuroblastoma cells. Thus, we investigated the mechanism of the neuroprotective effects of UA based on the results of Figs. 1 and 2.
The first mechanism of UA protection against neuronal apoptosis is inhibition of apoptosis through regulation of mitochondrial-related apoptosis pathways. Mitochondria play important roles in intracellular energy metabolism. Oxidative stress induce alteration in mitochondrial protein, lipids, and DNA. Structural damage of the mitochondria results in defects in mitochondrial function, that are connected apoptosis and caspase activation which in turn leads to the damage to nerve cell. Therefore, mitochondrial dysfunction has a crucial role in the pathophysiology of NDs [2542]. ROS-induced damage to mitochondrial membranes opens mitochondrial permeability transition pores (MPTPs), which induces release of mitochondrial cytochrome c. This activates caspase-9, which in turn triggers activation of caspase-3, leading to DNA damage, which impairs mitochondrial function [56]. During this process, members of the Bcl-2 family play important regulatory roles in the mitochondrial-related apoptosis pathway. Bax, a proapoptotic member, promotes apoptosis by accelerating the opening of MPTPs and inhibiting apoptosis via the anti-apoptotic Bcl-2 pathway [43]. In the present study, we confirmed increases in the Bax/Bcl-2 ratio, apoptotic nuclei, and expressions of cytochrome c, cleaved caspase-9, cleaved caspase-3, and cleaved PARP after H2O2 treatment. However, all of these apoptotic cell signals induced by H2O2 were significantly reduced by pretreatment with 2.5 and 5 µM UA (Figs. 3 and 4). Thus, UA inhibited apoptosis of SK-N-MC cells by inhibiting the mitochondrial-related apoptosis pathway. Tang et al. [18] reported that UA inhibits apoptosis of neonatal rat cardiomyocytes during hypoxia/reoxygenation (H/R) injury via the PI3K/Akt pathway. In addition, there have been many reports on the preventive competence and mechanism of EA, a metabolite precursor of UA. Firdaus et al. [44] reported that EA ameliorates mitochondrial dysfunction by reducing mitochondrial membrane potential and cytochrome c release against AS2O3-induced toxicity in SH-SY5Y human neuroblastoma cells. Chen et al. [45] reported that EA suppresses apoptosis by regulating the expressions of Bax, Bcl-2, and caspase-3 in the livers and brains of rats treated with D-galactose. In addition, Baluchnejadmojarad et al. [46] showed that EA has a protective effect via Nrf2/HO-1 signaling in a rat model of PD. These studies support our results that UA inhibits apoptosis. Therefore, we suggest that UA, which can pass through the BBB, may have a therapeutic effect on neurodegenerative diseases by ameliorating mitochondrial dysfunction of neuronal cells due to oxidative stress.
A second mechanism for the neuroprotective effects of UA involves modulation of the p38 MAPK pathway. MAPK pathways play an important role in cell proliferation, differentiation, and apoptosis regulation [47], and they are activated by various cell stress stimuli such as oxidative stress and endoplasmic reticulum stress [48]. The MAPK signaling pathway is involved in the pathogenesis of various diseases, including cancer and NDs. In the case of AD, activation of the MAPK pathway leads to neuronal apoptosis, β-secretase activity, γ-secretase activity, and tau phosphorylation [49]. In our study, pretreatment with UA significantly reduced expression of phosphorylated p38 MAPK (Fig. 5A). It also mitigated cell viability reduced by H2O2 via inhibition of the p38 MAPK signaling pathway (Fig. 5B). Although we did not confirm the effects of UA on phosphorylated ERK and the JNK pathway (data not shown), our results clearly indicate that UA protects cells from cytotoxicity by inhibiting activation of the p38 MAPK pathway. Chen et al. [50] showed that oral administration of EA induced downregulation of the JNK, p38 pathway and inflammatory mediators such as TNF-α, IL-1α, IL-1β, and COX-2 in induced hypoxic-ischemic brain-injury animal models. González-Sarrías et al. [51] reported that ≥ 10 µM EA and UA mitigated the inflammatory state in human colonic fibroblasts that induced inflammation by IL-1β via inhibiting the activation of NF-κB and the p38 MAPK pathway. In addition, Komatu et al. [16] reported that pretreatment with 40 µM UA inhibited the phosphorylation of p38 and JNK in inflammation-induced RAW264 macrophages induced by lipopolysaccharide, and also the formation of various proinflammatory mediators such as TNF-a, IL-6, and NO. In addition, p38 MAPK inhibitors are potential drug treatments for AD, and play an important role in the production of Aβ42-induced proinflammatory cytokines [52]. Therefore, we propose that UA inhibits the p38 MAPK pathway, which is an important mechanism for protecting neuronal cells and brain tissue through anti-apoptosis and anti-inflammatory effects.
In conclusion, UA inhibited apoptotic cell death involving oxidative stress by decreasing intracellular ROS production, inhibiting the mitochondrial-related apoptosis pathway, and modulating the p-38 MAPK pathway. Taken together, our results indicate that UA protects against H2O2-induced growth inhibition and apoptosis induction in SK-N-MC cells. Thus, we suggest that it could be a useful neuroprotective agent against oxidative stress-related brain diseases. Future studies should examine the metabolic processes of UA and its protective effects on brain tissue in animal models of AD and PD.
 XML Download
XML Download