

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Posterior cortical atrophy (PCA) is a rare degenerative disease with unknown cause. In 1988, Benson et al.1 reported some patients whose cerebral images showed significant atrophy of the parieto-occipital region with progressive dementia and cortical visual dysfunction. They named this disease as PCA based on image findings. Since PCA is most often associated with the pathological findings of Alzheimer's disease (AD), some people consider PCA as a visual variant of AD. Because PCA invades the posterior cerebral cortex, Gerstmann syndrome, Balint syndrome, and trans-cortical sensory aphasia may occur along with visual agnosia that occurs relatively early.23 Here we report a patient who visited our hospital with complaints of only visual symptom that rapidly progressed. We diagnosed it as PCA through clinical findings, image examinations, and other evidences.
CASE REPORT
A right-handed usually healthy 55-year-old female patient visited hospital with complaint of symptoms that she was not able to see well since about three months ago. She was a high school graduate. Although she changed her glasses, her symptoms were not improved. She even visited ophthalmology and received examination. However, no abnormal finding was discovered. Her visual field was shaken. She could not read newspaper or write in lines correctly. She sometimes ran into objects while walking. However, she had no big problem in finding her house.
The patient had nothing significant in her medical history. She had no family history of dementia. She did not show abnormal findings in physical examination. Her consciousness was clear. Due to her severe simultanagnosia, it was difficult to judge her accurate visual acuity or perform visual field examination. She was able to read small letters though. In addition, examinations on her cranial nerves, limb muscle strength and intensity, sensation, deep tendon reflex, and cerebellar function revealed normal findings. Her consciousness, attention, memory, and judgment all showed normal findings. She scored 26 on the Korean version of Mini-Mental State Examination. In Seoul Neuropsychological Screening Battery, agnosia of fingers was not observed. When her calculating ability was evaluated through mental arithmetic, she was able to do very simple addition and subtraction. However, her left and right orientations were reduced. For Rey Figure Drawing and Copying Test, she could not do painting over existing pictures or copy pentagon. When the letter '8' composing of the letter '3' was shown to her, she only mentioned small '3'. Therefore, optic ataxia and ocular apraxia were observed in addition to simultanagnosia. When she was asked to give answers to a number of colors, the patient could not answer most of them. However, when she was asked to identify whether they were the same color when two colors were presented, the patient answered accurately. When the pictures of celebrities or actors were shown, the patient could hardly recognize them. She even confused her husband with her boss at the company.
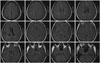
In laboratory tests including blood test, chemistry test, urinalysis, serum test, immune test, and cancer marker test, the patients showed normal findings. She did not show abnormal findings in electrocardiography, chest X-ray, or brainwave test. Because the patient showed too rapidly progressed visual symptoms, visual variant Creutzfeldt-Jakob disease (CJD) was suspected initially. However, she showed normal findings in diffusion weighted image. In addition, 14-3-3 protein was not observed in cerebrospinal fluid examination. In ApoE genotyping, the patient was found to be E4/E4 homozygote. No abnormal finding was observed in fluid-attenuated inversion recovery image (Fig. 1). In 18F fluoro-deoxyglucose positron emission tomography (18F FDG-PET), the FDG intake in the parietal-occipital lobe at both sides was significantly decreased (Fig. 2). With statistical parametric mapping analysis, hypometabolisms of the parietal lobe and occipital lobe at both sides as well as the left temporal lobe were identified (Fig. 3). Memory deterioration was unclear. We diagnosed it as PCA with remarkable visual symptom and image findings.
DISCUSSION
The patient visited our hospital with complaints of visual symptoms that occurred abruptly and progressed quickly in three months. In neuropsychological test, findings implied damages of parietal lobe dysfunctions at both sides with optic ataxia, simultanagnosia, reduced visuo-spatial dysfunction, reduced left and right orientations, and acalculia. In addition, prosopagnosia implying lesion of the parietal temporal lobe was observed. In PCA, unlike typical AD, memory and insight will be relatively maintained until the end of the disease. In addition, there will be no personality change or abnormal behavior. Furthermore, Gerstmann syndrome occurs much earlier with frequent Balint syndrome.1 Our patient showed very similar clinical patterns to those of patients with PCA reported previously.4567 However, her central visual dysfunction and visuo-spatial dysfunction were significant without clear memory impairment. This might be due to the fact that the examinations were conducted relatively early. Considering the fact that the progression of visual symptoms was very fast, differentiation from other diseases might be needed.
Kennedy8 reported that hypometabolism was remarkable in the parietal lobe and temporal lobe association cortex. In FDG PET of this patient, hypometabolisms in the parietal-temporal lobe regions at both sides were observed, which could be distinguished from typical AD. Although dementia with Lewy bodies (DLB) and CJD in the form of Heidenhain has shown hypometabolism in the occipital lobe in FDG PET,910 it can be distinguished from DLB through the changes in the level of consciousness and movement symptoms of Parkinson's disease. In addition, this disease can be distinguished from CJD through quickly progressed dementia, periodic sharp wave complexes in brainwave test, and presence or absence of 14-3-3 protein in cerebrospinal fluid examination.11
Signs of PCA have been reported in patients with CJD in some forms of Pick's disease and Heidenhain.7 As mentioned above, patients with PCA can relatively maintain memory until the end of the disease. They have conscious reaction to neuropsychological test and they attempt to participate in it. However, patients with AD, CJD, and Pick's disease will lose understanding early. Alexia and agraphia are relatively early symptoms of PCA when compared to AD or Pick's disease. In addition, paramyoclonus sometimes can occur in the latter period of PCA, which is different from CJD that occurs much earlier.12 Currently, as many studies on AD have been conducted and various drugs have been developed, the importance of early diagnosis of AD is being emphasized. It is also important to have a quick and accurate diagnosis of PCA, a visual variant AD. Through this case, progressive visual dysfunction can be shown at a relatively young age. Therefore, PCA should be included as a differential diagnosis of AD although memory impairment is not always accompanied. Neuropsychological test and neuroimaging tests including FDG PET should be conducted and analyzed to differentiate PCA from AD.


 XML Download
XML Download