

 PDF
PDF ePub
ePub Citation
Citation Print
Print



INTRODUCTION
Breast cancer is the most diagnosed type of cancer in women, and it has the second highest mortality rate in this group, following only lung cancer [1]. Triple-negative breast cancer (TNBC) defines a subtype of breast cancer that does not express estrogen receptor (ER), progesterone receptor, and human epidermal growth factor receptor 2 (HER2). Approximately 20% of breast cancers in women are TNBC, and this type is more aggressive and often diagnosed in younger patients [23]. It is a clinical challenge to treat TNBC, as no therapeutic targets have so far been successfully described, and its prognosis remains modest.
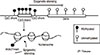
Different research groups have proposed molecular classifications based on gene expression profiling (Figure 1). Thus, five intrinsic subtypes of breast cancer have initially been defined [45]. Subsequently, in order to translate these findings and make them suitable for clinical application, immunohistochemical equivalents to the intrinsic subtypes have been established [67]. Among these subtypes of TNBC, approximately 80% are basal-like [89]. However, not all basal-like breast cancers are triple-negative, and up to 20% of basal-like tumors are either ER+ or HER2+ [8]. Other subtypes include the claudin-low [10] and the interferon-rich, which is closely related to TNBC [11].
Most gene expression profiling studies on TNBC have been conducted in order to establish better prognostic or predictive factors. One of the first studies by Lehmann et al. in 2011 described six subtypes: two basal-like (BL1 and BL2), one immunomodulatory (IM), one mesenchymal (M), one mesenchymal stem-like (MSL), and one luminal androgen receptor (LAR) [12]. The MSL and IM subtypes were subsequently removed because they are formed by tumor-associated stromal cells and infiltrating lymphocytes, respectively [13] (Figure 1). Burstein et al. [14], in 2015, described four subtypes with clinical significance: luminal-AR (LAR), mesenchymal (MES), basal-like immune-suppressed (BLIS), and basal-like immune- activated (BLIA). Among these, BLIA and BLIS had the best and worst prognoses, respectively, according to both disease-free and disease-specific survival parameters. Moreover, subtype-specific targets were identified [14]. Another study identified three clusters of TNBC by microarray profiling: luminal androgen receptor (C1), basal-like with low immune response and high M2-like macrophages (C2), and basal-enriched with high immune response and low M2-like macrophages (C3) [15], with the better outcome being attributed to the C3 cluster. A review of currently developed systems of classification for TNBC is provided by Ahn et al. [16].
These findings all suggest that immunohistochemically defined TNBCs generally display basal-like properties when gene expression is analyzed. In this review, we will focus on discoveries that have been made regarding TNBC, including its basal-like equivalent, as there is an 80% overlap between the two entities [89]. However, it is important to mention that these two terms are not synonymous, as they both define molecularly heterogeneous entities.
EPIGENETIC MODIFICATIONS: AN OVERVIEW
Considering the heterogeneity of TNBC, current research is focused on finding new approaches to target this neoplasia; one such strategy employs the use of epigenetics. Being increasingly recognized in all types of cancer, epigenetics plays a major role in tumorigenesis. This field is defined as the study of heritable changes in gene expression without alteration in DNA sequences [17]. Multiple epigenetic modifications with diagnostic, prognostic, or therapeutic significance have already been reported in a number of malignancies, including breast cancer [18].
The first and main epigenetic modifications described and accepted by a large number of authors are DNA methylation and posttranscriptional modifications of histones [171920]. Other more recently described and accepted modifications are noncoding RNAs (ncRNAs) [171819], as well as chromatin remodeling [1920], nucleosome positioning, and chromosomal looping [17]. All these markers are strongly interconnected, and one epigenetic modification can easily induce another, as shown in certain circumstances presented later in the article.
DNA methylation is one of the most well-described epigenetic events. Cytosine methylation in CpG islands is a recognized marker of epigenetic silencing and is performed by DNA methyltransferases (DNMTs), among which DNMT1 is responsible for maintaining methylation patterns following replication, and DNMT3a and DNMT3b initiate de novo methylation [17].
Histone modifications are covalent posttranslational alterations to histone proteins that influence the chromatin structure and consequent gene transcription; hence, they are important epigenetic markers. Histone modifications include methylation, phosphorylation, acetylation, ubiquitylation, and sumoylation, and they can either activate or deactivate gene expression. Some specific modifications have been correlated with carcinogenic events. Methylation of histone H3 at lysine residues 9 and 27 (H3K9me3, H3K27me3) by the polycomb repressor complex 2 (PRC2) is a hallmark of silenced chromatin [20]. Specific modifications such as lysine acetylation (H3K9ac, H3K18ac, and H4K12ac), lysine trimethylation (H3K4me3), and arginine dimethylation (H4R3me2) are recognized markers of gene activation; on the other hand, lysine methylation (H3K9me2 or H3K9me3 and H4K20me3) is usually associated with gene silencing [21].
ncRNAs do not encode protein, but rather modulate chromatin regulation and gene expression [17]. They include transfer RNAs (tRNAs), ribosomal RNAs (rRNAs), small nucleolar RNAs (snoRNAs), and the recently discovered and largely studied long noncoding RNAs (lncRNAs) and microRNAs (miRNAs) [22]. Both lncRNAs and miRNAs are widely studied and have been strongly linked to a large number of diseases.
For didactic reasons, this article has been further divided according to the most relevant types of epigenetic modifications, namely DNA methylation, noncoding RNAs, and histone modifications, as these are the most well-described and widely studied (Supplementary Table 1, available online). However, all these modifications are strongly interconnected, and the regulation of one gene may be the product of more than one epigenetic modification.
DNA METHYLATION
Several studies have looked into the DNA methylation patterns in various breast cancers, including TNBC and its closely related basal-like counterpart.
DNA methylation in triple-negative breast cancer
One of the most comprehensive analyses of the TNBC methylome stratified patient samples into three methylation clusters based on differentially methylated regions (DMRs) [23]. The hypomethylated profile was associated with better survival within the first 5 years post-diagnosis compared with the more heavily methylated subtypes, while the medium methylated cluster was associated with the worst survival. It also identified 17 individual DMRs capable of stratifying TNBC patients into good and poor prognosis groups. Among the genes included are the WT1 gene and its antisense counterpart, WT1-AS, for which high levels of methylation correlated with elevated levels of expression and poor survival. Hypermethylation of the bi-directional promoter is associated with decreased WT1 and WT1-AS expression and improved survival; however, these findings remain to be verified on a larger cohort [23]. The study also described hypermethylation events to mostly occur in CpG islands in the context of global hypomethylation (Figure 2). The hypermethylated regions correlated strongly with the regions of human mammary epithelial cells marked with H3K27me3, a marker of epigenetic silencing. Specifically, 12 methylated genes were identified as both mutated and downregulated; these included ROBO3 and SEMA5A [23], which are genes involved in axon guidance, a pathway that has been newly implicated in tumor initiation and progression in breast cancer [24]. This pathway, originally described in brain development [25], includes the Slit, Netrin, Eph/ephrin, and Semaphorin proteins, which have recently been found to regulate normal mammary development, as well as breast cancer initiation, progression, and angiogenesis [26]. Promoter hypermethylation was found in seven members of this pathway, which may prove to be promising for future investigations in targeted cancer therapy [23].
An earlier study also described a specific methylation pattern for TNBC by analyzing the hypermethylation of 110 CpG islands in 69 cancer-related genes. The TNBC-specific profile was defined by the methylation of five genes (CD44, MGMT, CDKN2B, RB, and p73) and the non-methylation of 11 genes (GSTP1, PMS2, MSH2, MLH1, MSH3, MSH6, DLC1, CACNA1A, CACNA1G, TWIST1, and ID4), with MGMT, MMR, and ID4 showing the strongest association [27]. Interestingly, there was no significant difference in the methylation of the BRCA1 and BRCA2 promoters between triple-negative and non-triple-negative tumors. However, ID4, one of the genes in the non-methylated group, is a negative regulator of BRCA1; this may imply a new mechanism of BRCA silencing that is worth investigating.
DNA methylation and BRCAness in triple-negative breast cancer
Up to 30% of TNBC cases have a BRCA mutation [28], and there is a strong association between the two entities, usually leading to a poorer prognosis [29]. However, a large number of tumors share the molecular features of BRCA-mutant cancer, a state defined as “BRCAness” [30]. This particular status may be due to the hypermethylation of the promoter region of the BRCA1 gene [313233]. There seems to be a mutually exclusive relationship between a BRCA1 mutation and promoter methylation [31]. Moreover, TNBC with BRCAness may not only benefit from therapy with poly (ADP-ribose) polymerase (PARP) inhibitors and platinum agents [3435], but also show a survival benefit from anthracycline-based chemotherapy [36].
DNA methylation and triple-negative breast cancer progression
Another whole genome methylation analysis compared the primary tumor to normal adjacent tissues and lymph node metastases, and identified a set of aberrations that may explain the progression of TNBC [37]. Sixteen genes that were identified to be specific to TNBC also had differentially methylated probes, including five genes classified as DMRs–ANKRD30B, COL14A1, IGF1, IL6ST, and MEG3. An additional set of genes were found to be differentially methylated in the lymph node metastases. Some of these genes correlated with better survival; particularly, the higher methylation of SPRY2, EGR1, GREB1, ITIH5, and LRRC17, and the low methylation of AMIGO2. The same study found that EGR1 downregulation is inversely correlated with its methylation [37]. Furthermore, a specific gene, BRMS1, may epigenetically influence the metastatic potential of TNBC [38]. The expression of BRMS1 was found to be significantly reduced in TNBC tissue samples and cell lines when compared to normal breast tissue; it was also inversely correlated with lymph node metastasis. DNA methylation-dependent inactivation was proven on breast cancer cell lines (MDA-MB-231, HCC-1937, and MDA-MB-435), a normal breast tissue cell line (MCF-10A), and on primary breast cancer tissues with matched nonmalignant breast tissue [38]. Methylation of this gene significantly correlated with larger size and higher tumor-node-metastasis (TNM) stage of the tumor, suggesting that this gene may function as a tumor suppressor.
DNA methylation and cancer stem cells in triple-negative breast cancer
The role of DNA methylation in TNBC was elucidated by investigating the regulation of breast cancer stem cells (CSC) through promoter methylation [39]. One study found that the promoter regions of CD44, CD133, and Musashi-1 (MSI1), which are genes associated with stem cell properties [39], were hypomethylated in primary breast cancer samples, and this correlated with a TNBC subtype and a clinically aggressive phenotype.
DNA methylation in basal-like breast cancer
In a study by The Cancer Genome Atlas Network, primary breast cancer tissues were analyzed using multiple platforms, including DNA methylation, exome sequencing, messenger RNA (mRNA) arrays, and miRNA sequencing [40]. There was a high degree of overlap between basal-like and TNBC-defined samples, and the basal-like subtype clustered together most distinctively across all platforms. The study described a hypomethylated phenotype of basal-like tumors, and their findings showed that this subtype correlated with the lowest levels of DNA methylation and up to 80% frequency of TP53 mutations, as well as being frequently associated with loss of RB1 and BRCA. Interestingly, these findings suggest that basal-like tumors are similar to serous ovarian carcinomas, raising the hypothesis that common therapeutic approaches should be considered [40]. Moreover, the frequency of BRCA1 and BRCA2 mutations in these tumors is similar to that of TNBCs, confirming that these subtypes may benefit from PARP inhibitors and platinum compounds.
Specific DNA methylation patterns in basal-like tumors have also been reported in comparison to that in luminal A and B tumors in a study that investigated the methylation profiles of the five intrinsic subtypes of breast cancer [41]. RASSF1 and GSTP1, genes usually associated with the ER+ phenotype [42], were unmethylated in basal-like tumors, in contrast to that of the luminal B phenotype; on the other hand, ARHGDIB, GRB7, and SEMA3B were found to be significantly more methylated in basal-like tumors [41]. Another significant difference observed between BRCA mutation carriers is that BRCA2 tumors were significantly more methylated than BRCA1 tumors. Overall, the basal-like phenotypes had lower overall methylation than the other subgroups [41], which supports the findings of the aforementioned studies.
A hypermethylator phenotype has also been described for basal-like breast cancer, namely the CpG island methylator phenotype [2343]. This does not refer to global hypermethylation but describes concurrent methylation-dependent silencing of a number of genes, including a specific set of genes with predictive power (CDH1, CEACAM6, CST6, ESR1, LCN2, and SCNN1A) that are involved in a wide range of malignancies [44]. This specific pattern of methylation may be linked to the overexpression of DNMT3b. Moreover, DNMT3b seems to be a promising target for TNBC, as shown by the targeted inhibition of DNMT3b by RNAi-mediated knockdown in three cell lines (MDA-MB-453, BT549, and Hs578T); all cell lines subsequently showed increased sensitivity to doxorubicin, paclitaxel, and 5-fluorouracil [45].
DNA methylation is one of the most well-studied epigenetic events, and it has also been well-studied in TNBC. However, findings on methylation patterns need to be translated into clinical practice, either by using this data to stratify patients' prognoses and expected outcomes [23273740] or by further investigating the pathways that have been indicated to show promising results [23314045].
NONCODING RNAs
Long noncoding RNAs in triple-negative breast cancer
A novel classification scheme for TNBC was established by Liu et al. [46] by integrating the profiles of mRNAs and lncRNAs. Four distinct clusters have been described—IM, LAR, MES, and BLIS—and these partly correlated with the Lehmann subtypes that have been described before [9]; furthermore, the BLIS subtype has been described as the most aggressive phenotype [46].
Further microarray profiling of TNBC has identified a number of lncRNAs with different expression patterns compared to normal tissue [47]. However, the functions, pathway interactions, and importance of these remain to be established. Similarly, another microarray profiling study of lncRNAs in TNBC patient tissue samples found that the dysregulation of the ER in TNBC may be associated with lncRNA LINC00993 [48]. Recently, another lncRNA, MALAT1, was found to play a role in the metastatic potential of TNBC and is reported as a potential prognostic marker for lymph node-negative HER2+ and TNBC [49].
Long noncoding RNAs in basal-like breast cancer
lncRNAs are ncRNAs longer than 200 nucleotides and have various functions in the genome [22]. The expression of lncRNAs in breast cancer has also been investigated using The Cancer Genome Atlas project database [50]. Four clusters were described, with the first being almost entirely populated by the basal-like subtype. HOTAIRM1 was found to be overexpressed in this cluster; however, its role still remains to be determined [50].
Another promising lncRNA target is FOXCUT, which may be a cancer-promoting gene responsible for the aggressive phenotype of basal-like tumors. A study has reported that FOXCUT was significantly more highly expressed in basal-like than in non-basal breast cancer subtypes, and that its knockdown inhibited cell migration and proliferation. These data show that FOXCUT may be a potential diagnostic and therapeutic marker of basal-like TNBC [51].
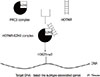
A lncRNA linked to aggressive progression in breast cancer through H3K27 methylation [52], HOTAIR, has been found to be upregulated in MCF-7-TNR cells, the basal-like derivative of the luminal-like MCF-7 cells. HOTAIR and its partner enhancer of zeste homolog 2 (EZH2) form a complex that seems to play a critical role in the maintenance of the basal-like phenotype; when either HOTAIR or EZH2 was inhibited, the dysregulated expression of luminal-like and basal-like markers was attenuated and the proliferation of MCF-7-TNR cells was inhibited (Figure 3). HOTAIR is also required for the expression of basal-like genes and the proliferation of MDA-MB-157 cells [53]. Furthermore, it was shown that co-targeting EGFR and c-ABL in TNBC cell lines through lapatinib and imatinib, respectively, inhibited growth by downregulating HOTAIR [54].
MicroRNA in triple-negative breast cancer miRNAs are small ncRNAs approximately
miRNAs are small ncRNAs approximately 20 nucleotides long that can posttranscriptionally alter gene expression [22]. Gasparini et al. [55] identified a four-miRNA signature in TNBC that allowed the stratification of patients into high- and low-risk groups. Upregulation of miR-493 and miR-155 correlated with better patient outcome, whereas downregulation of miR-30e and miR-27a correlated with a negative outcome [55].
miRNAs have also been described to be potential TNBC biomarkers. miR-10b, miR-26a, miR-146a, and miR-153 were investigated in breast cancer cell lines and were linked to BRCA1 expression. In MDA-MB-231 cells, BRCA1 expression is downregulated by miR-10b and miR-26a. miR-146a is significantly overexpressed in TNBCs without affecting the expression of BRCA1, while miR-153 can upregulate BRCA1 expression in MDA-MB-231 cells [56]. However, Kumaraswamy et al. [57] reported that BRCA1 expression positively correlates with miR-146a and leads to the downregulation of EGFR. Furthermore, Garcia et al. [58] reported that miR-146a and miR-146b-5p downregulate BRCA1 in TNBC. In a study by Murria et al. [32], miR-590-5p and miR-4417 were found to be hyperexpressed in TNBC. miR-590 can impact ER regulation by interacting with the two mRNA sequences of ESR1, while miR-4417 can regulate BRCA1 mRNA [32].
miRNAs are also drivers of epithelial-to-mesenchymal transition (EMT), an important process in initiating metastasis. An insight into the mechanism that controls their expression in TNBC and correlation to node metastasis was provided by a recent study in which the interaction between two epigenetic mechanisms was shown. miR-200c/miR-141 locus methylation is associated with low miR-200c expression and lymph node invasion in TNBC, favoring metastasis and altering TNBC prognosis [59]. This has also been associated with high levels of ZEB1 transcription factor, which is involved in EMT, suggesting the miR-200c/ZEB1 axis as a possible therapeutic target in metastatic TNBC. Moreover, the mir-200 family of miRNAs has been shown to play important roles in TNBC. Ectopic expression of miR-200b suppressed TNBC migration and metastasis in a mouse mammary xenograft tumor model by inhibiting protein kinase Cα [60]. Another member, miR-200a, has also been shown to modulate TNBC migration by regulating the EPHA2 oncogene [61], while overexpression of miR-200b-3p and miR-429-5p inhibits the proliferation, migration, and invasion of TNBC cells by inhibiting the LIMK1/CFL1 (LIM domain kinase 1/cofilin 1) pathway [62], thus opening new possibilities for targeted therapies in TNBC.
A comprehensive summary of miRNAs with profiling, functional, prognostic, and therapeutic potential is provided by Mathe et al. [63].
HISTONE MODIFICATIONS
Histone modifications in triple-negative breast cancer
Eight key histone modifications—H3K4me1, H3K4me3, H3K9me3, H3K9ac, H3K27me3, H3K27ac, H3K36me3, and H3K79me2—have been profiled across 13 cell lines, including four TNBCs—MDA-MB-231, MDA-MB-436, MDA-MB-468, and HCC1937 [64]. Subtype-specific histone modification profiles have also been discovered, including distinct H3K36me3 patterns in TNBC cell lines. The androgen receptor (AR) pathway genes were active especially in claudin-low TNBC cell lines, while AR pathway regulators had lower expression levels in basal-like cells lines [64]. Another specific TNBC chromatin state that was identified is the AFAP1-AS1 marked by the active H3K4me3 and H3K79me2 modifications. The authors reported that this gene has not been linked to TNBC before but is highly expressed and predicts poor prognosis in other cancers, including esophageal adenocarcinoma, pancreatic ductal adenocarcinoma, lung cancer, nasopharyngeal carcinoma, hepatocellular carcinoma, and colorectal cancer; it may also promote tumor invasion by EMT. Small interfering RNA mediated depletion of AFAP1-AS1 in MDA-MB-231 and HCC1937 cells led to decreased proliferation and colony formation [64].
A transcription factor that has been recently characterized, BCL11A, is overexpressed in TNBCs, including basal-like subtypes [65]; it is important for mammary stem and progenitor cells [65] and it promotes tumor formation by interacting with a common subunit (RBBP4/7) of the histone methyltransferase (PRC2) and histone deacetylase (NuRD, SIN3A) complexes [66] to regulate transcription and promote tumorigenesis.
Another family of proteins involved in the epigenetic regulation of gene expression is the bromodomain and extra-terminal (BET) family; they recognize acetylated lysine residues in nucleosomal histones [6768]. Inhibition of these proteins has been shown to exhibit antitumoral efficacy in solid tumors, including TNBC [68697071]. Many BET inhibitors have shown promising results in preclinical research studies, including synergistic effects with already established therapies [67707273] and the compound OTX015/MK-8628, is in clinical development for TNBC [67].
Histone modifications seem to play an important role in the EMT of TNBC, as reported by a study using the basal-like cell line, MDA-MB-231. The downregulation of histone methyltransferase G9a, histone acetyltransferase KAT5, and H3K79 methylator DOT1L induce E-cadherin expression and promote an epithelial phenotype with lower migratory and invasive capacity [74]. These findings may prove to be useful insights into using epigenetic targets as means of reducing the risk of metastasis. EMT and mesenchymal state maintenance may also be influenced by a histone 2 variant, macroH2A1. Overexpression of macroH2A1.1 correlated with mesenchymal markers of the claudin-low breast cancer subtype and with poor prognosis in TNBCs [75].
Histone modifications in the basal-like subtype
A histone modification profile specific for breast cancer subtypes was generated from a series of 880 human breast carcinomas [21]. Moderate to low levels of lysine acetylation (H3K9ac, H3K18ac, and H4K12ac), lysine methylation (H3K4me2 and H4K20me3) and arginine methylation (H4R3me2) were reported in carcinomas of poor prognostic subtypes, including basal carcinomas. However, even if the basal-like carcinomas were represented in the low histone modification cluster, the HER2-positive cancers had even lower frequencies of histone modifications [21]. Similar findings were described with regard to a single histone alteration, H3K27me3, that is inversely associated with HER2-positive and basal-like breast cancers [76]. In the latter, H3K27me3 seems to be mediated by a higher expression of EZH2, a member of PRC2, leading to histone-mediated silencing of PRC2 target genes [41].
Breast cancer stem cells (BCSC) being important drivers of TNBC aggressiveness is supported by a new study by Li et al. [77], wherein they sorted a population of CD44+/CD24− BCSCs from a culture of MDA-MB-231 cells. Their findings showed that both DNA and histone methylation differed between CSCs and non-CSCs. In particular, H3K4me2 and H3K27me3 were both decreased in CSCs and may have affected both Wnt and GnRH signaling. The sorted CSCs demonstrated greater invasive and tumorigenic capacities both in vivo and in vitro [77]. However, the exact mechanisms underlying these remain to be elucidated.
Therapeutic potential of histone modifications
Even if histone modification mechanisms in TNBC are still not fully understood, therapies based on these hallmarks already show promising results in preclinical studies. Some of the most widely used epigenetic therapies are based on histone deacetylases (HDACs). These enzymes remove acetyl groups from histones and are thus responsible for regulating gene expression, including tumor suppressors. HDAC inhibitors (HDACi) are currently being investigated in a large number of solid and hematological malignancies, and they have been shown to inhibit tumor growth and induce apoptosis by targeting multiple pathways [78]. A study has shown that HDACi suberoylanilide hydroxamic acid (vorinostat) and sodium butyrate inhibit cell proliferation, induce apoptosis, and downregulate transcription of mutant p53 in TNBC cell lines MDA-MB-231 and BT-549 [79]. Targeting p53 is a strategy that has been successfully investigated in TNBC [808182]. Similar results have been found for HDACi with panabinostat, which has been shown to induce hyperacetylation of histones H3 and H4, decrease proliferation and survival, and induce apoptosis in TNBC cell lines MDA-MB-157, MDA-MB-231, MDA-MB-468, and BT-549. Panabinostat also decreased tumor size in vivo in mice models for the MDA-MB-231 and BT549 lines [83]. Vorinostat enhanced the growth inhibitory ability of PARP inhibitor olaparib in TNBC cells with overexpressed PTEN, while PTEN knockdown cells were resistant to this combination. The results were confirmed in an in vivo MDA-MB-231 mouse model [84].
HDACi may aid in targeting the PD-1/PD-L1 pathway, which regulates T cell function [85]. Several breast cancer cell lines, including TNBC MDA-MB-231, were treated with both class nonspecific (vorinostat and panobinostat) and specific HDACi (valproic acid and entinostat), leading to the upregulation of PD-L1 on tumor cells [86]. A combination of vorinostat and immune checkpoint inhibitors (PD-1 and CTLA-4 blockade) on mice models of TNBC led to decreased tumor growth and prolonged survival. The authors described that vorinostat also promoted Treg downregulation in vitro and increased T cells tumor infiltration in vivo. This data suggests that HDACi potentiates immune checkpoint inhibitor blockade in TNBC [86].
EMT and metastasis become irreversible when a subpopulation of tumor cells gains the ability to spread from the primary tumor and establish secondary localizations. HDACi entinostat reduces the expression of markers associated with this cell population in TNBC cell lines MDA-MB-231, BT549, and Hs578T; decreases the ability of MDA-MB-231 to form lung metastasis in an in vivo mouse model; and reduces tumor formation from patient-derived xenografts [87]. Furthermore, vorinostat also has the ability to prevent brain metastasis of TNBC in vivo [88], proving that HDACi should be further investigated for use in the management of metastatic TNBC. Finally, Mekala et al. [89] suggested that HDACi may also aid in re-expression of miRNAs and, by regulating the miR-200 family through HDACi, open a new avenue for research in TNBC.
Other histone-modifying enzymes that can promote aggressiveness of TNBC are histone methyltransferases. A histone methyltransferase, hSETD1A, has been associated with poor outcome and decreased overall survival rates in a retrospective study on 159 TNBC patients [90], indicating that it could be further investigated as a prognostic marker.
OTHER EPIGENETIC CHANGES
These epigenetic changes have an important impact on how genomic DNA is organized, either into tightly packed heterochromatin or as loosely packed euchromatin. DNA methylation, histone modifications, or ncRNAs may recruit protein complexes that indirectly regulate gene expression by how much access to DNA they allow transcription machinery.
Chromatin remodeling refers to the regulation of gene expression by modifying the chromatin architecture to either allow or restrict transcription. This can be done either by post-transcriptional modifications of histones or ncRNAs and their recruitment of the PRC2 complex as previously mentioned, or by ATP-dependent chromatin remodeling protein complexes [19]. SWI/SNF is one of the most well-described complexes with this function [19]. Two ATPases of this complex, BRG1 and BRM, have elevated levels in breast cancer, and their knockdown in a TNBC cell line led to reduced tumor formation in vivo and reduced cell proliferation in vitro [9192]. In addition, knockdown of BRG1 sensitized a TNBC cell line to doxorubicin, 5-fluorouracil, gemcitabine, cisplatin, cyclophosphamide, and paclitaxel [93].
CONCLUSION
TNBC is a heterogeneous oncological entity for which, even if major breakthroughs have been made to describe subtypes with relevance in clinical practice, no specifically designed tool for management exist to date. Epigenetic modifications are currently being intensely studied in all malignant diseases, including breast cancer. TNBC may especially benefit from advances in this domain, considering that no therapeutic targets currently exist for this subtype. We now have at our disposal a multitude of methods to study these epigenetic machinery, and a large amount of data is constantly being generated. Several studies employing epigenetic drugs, namely HDACi, already show promising results. One particular drug, tinostamustine, is a first-in-class alkylating deacetylase inhibitor that combines the DNA-damaging effect of bendamustine with the HDACi vorinostat in a completely new chemical entity [94]. Tinostamustine is currently being tested in a phase I/II clinical trial that also enrolls TNBC patients (NCT03345485). However, the main challenge in translating data into clinical practice still remains. A targeted approached based on identifying which mechanisms drive TNBC in the absence of known receptors and which mechanisms are responsible for its aggressiveness may prove to be a more efficient method for developing new treatments and markers. This malignancy would especially benefit from better classification tools that can identify the patients who can benefit from a certain treatment. The epigenetic field is a very promising area where such answers may be found.

 XML Download
XML Download