

 PDF
PDF ePub
ePub Citation
Citation Print
Print


| Ann Clin Neurophysiol. 2017 Jan;19(1):3-12. English. Published online Jan 26, 2017. https://doi.org/10.14253/acn.2017.19.1.3 | |
| Copyright © 2017 The Korean Society of Clinical Neurophysiology | |
|
David Burke, | |
|
1Department of Neurology, Royal Prince Alfred Hospital, University of Sydney, Sydney, Australia. | |
|
2Brain and Mind Centre, University of Sydney, Sydney, Australia. | |
| Received September 06, 2016; Accepted September 15, 2016. | |
|
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by- | |
|
Abstract
| |
|
Using threshold tracking, differences have been established between large myelinated sensory and α motor axons in humans. Major differences are that sensory axons are relatively depolarised at rest such that they have a greater persistent Na+ current, and have greater activity of hyperpolarisation-activated cyclic nucleotide-gated (HCN) channels. Sensory axons may thereby be protected from hyperpolarising stresses, and are less likely to develop conduction block. However, the corollary is that sensory axons are more excitable and more likely to become ectopically active. |
|
Keywords: Axonal excitability; Threshold tracking; Sensory axons; Motor axons; Conduction block |
|
|
INTRODUCTION
|
Axons are required to maintain specific discharge rates and patterns, and as a consequence, the ability to conduct an impulse with minimal expenditure of energy will create different needs for different axonal populations. There may be only one role for an axon, and that is to conduct an impulse securely from one end to the other, but it is to be expected that the biophysical properties of sensory and motor axons may differ in order for them to fulfil this role. Specifically, motor axons that innervate muscle and cause it to contract will behave differently to sensory axons that arise from skin (or muscle) and provide feedback to the central nervous system.
Axons are more than telecommunication cables. Were it not for the investment of channels and pumps and the specific organisation of myelin, which in turn determines the localisation of those channels and pumps, the message would decay with distance in accordance with the “cable properties” of the axon. The present Review will consider the basis for the differences in properties of large sensory and motor axons and how these differences influence their behaviour in neurological disease.
|
CONSIDERATIONS WHEN STUDYING HUMAN SUBJECTS IN VIVO
|
In human studies threshold-tracking techniques enable the study of axonal behaviour under sufficiently controlled conditions to make inferences about some of the underlying biophysical processes. In these studies membrane potential can be altered using depolarising or hyperpolarising currents, to enable conclusions to be reached about the mechanisms underlying the resultant changes in excitability, particularly voltage-dependent processes. It should be kept in mind that precisely the same mechanisms may not be responsible for a property when disease alters the structure and function of an axon. This is in part because the distribution of different ion channels along an axon is largely determined by the nodal apparatus and the myelin sheath, is not homogeneous and can change in disease states, and channel isoforms that are not present in healthy nerve can then appear.
|
ION CHANNELS AND RELATED AXONAL PROCESSES
|
The Na+ channel is the key axonal channel for impulse generation. These channels have a high density at the node of Ranvier (Fig. 1), and thereby provide the substrate necessary for saltatory conduction. They are also present on the internode, though in a density that is about one thirtieth of that at the node.1 As a result, the internodal membrane undergoes changes in excitability during and after an action potential. These fluctuations in internodal membrane potential are insufficient to generate an “internodal action potential” unless there is a redistribution of Na+ channels normally present at the node (Nav1.6) and isoforms that are not active in the normal adult axon appear (e.g., Nav1.2), as may occur with chronically demyelinated lesions in multiple sclerosis.2 The Nav1.6 isoform can have two gating modes: approximately 98-99% of the channels are rapidly inactivated with maintained depolarisation (thereby passing a “transient” current), and this prevents them from passing further current which would increase the depolarisation and thereby lead to greater channel opening. However 1–2% of channels do not inactivate, or do so very slowly (and are thereby responsible for a “persistent” current). They are activated at less depolarised membrane potentials and some may be open at rest. They have a destabilising influence on membrane potential, and this needs to be balanced by measures that extrude Na+ ions and thereby limit the depolarisation. The current through Na+ channels that do not inactivate is referred to as “persistent” Na+ current, /NaP.
|
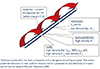
K+ channels with fast kinetics (the so-called “delayed rectifier”) are largely sequestered in the juxta-paranodal region under the myelin sheath (Fig. 1), such that access to them is restricted provided that the paranodal seal is intact.1 With paranodal changes, this restriction may be impaired so that in some disease states fast K+ channels can have actions that are denied them in healthy axons. However there are K+ channels at the node:1, 3 those with slow kinetics are present in a density 30 times greater than that on the internodal membrane, and approximately 35% are open at resting membrane potential, at least in the rat. This means that they can exert a hyperpolarising influence on the fluctuations in excitability following an action potential. Because a significant percentage of nodal slow K+ channels are already open at rest, they help determine the duration of the relative refractory period in healthy axons, while fast K+ channels cannot, unless the paranodal seal is impaired.
Based on pharmacological evidence,3 “hyperpolarisation-activated cyclic nucleotide gated (HCN)” channels are located primarily on the internodal membrane. These channels have very slow kinetics and are activated by hyperpolarisation, more so the greater the hyperpolarisation. They pass an inward current, /h, which depolarises membrane potential and counteracts the hyperpolarisation, thereby returning membrane potential closer to rest. This accommodative change to the hyperpolarisation is commonly termed inward rectification.
The Na+/K+ pump is an energy dependent process that consumes ATP. It is the prime mechanism for restoring Na+ balance on either side of the axonal membrane and a major determinant of resting membrane potential. The operation of the pump is responsible for much, if not most, of a neuron's energy expenditure. The pump is active at rest and contributes 10–15 mV to resting membrane potential.4 It extrudes three Na+ ions and brings two K+ ions into the axon, an imbalance that hyperpolarises the membrane. The pump is therefore termed “electrogenic”. Accordingly paralysis of the pump by ischaemia (removing its ATP source) results in axonal depolarisation, and on release of ischaemia axons will hyperpolarise as the pump resumes activity and attempts to lower the raised intra-axonal Na+ concentration. Another important mechanism that can alter intra-axonal Na+ concentration is the Na+/Ca2+ exchanger, which is co-localised with Na+ channels2 and normally functions to minimise intra-axonal Ca2+ concentration by exchanging Ca2+ ions for Na+ (three Na+ for one Ca2+). However with depolarisation the exchanger can operate in reverse mode, and this occurs at the peak of the action potential, when Na+ influx is greatest.5 It also occurs during ischaemia. Reverse operation results in Ca2+ ions being brought into the axon in exchange for Na+. Theoretically at least the conduction of prolonged impulse trains by ischaemic axons could result in a potentially damaging increase in intracellular Ca2+ concentration and trigger “excitotoxicity”.6
Although the action potential is generated at the node of Ranvier, the excitability of the axon is strongly influenced by the properties of the internode. This is because there are many more channels on the internode than the node, which is much smaller. Specifically, the number of Na+ channels is higher on the internodal membrane than on the nodal membrane, even though channel density is thirty times greater at the node. Thus the action potential rides on a background excitability: whether it will occur depends on whether the driving Na+ current can raise membrane potential to threshold. Theoretically then conduction block can be produced by decreasing the action current or by hyperpo-larising the axon, and this will be examined further, below, for human axons.
|
DIFFERENCES BETWEEN SENSORY AND MOTOR AXONS
|
The biophysical properties of sensory and motor axons differ, largely because of adaptations to their rate and pattern of discharge. It is a common clinical finding that a disease process seems to target a specific axonal population: for example, there is greater susceptibility to paraesthesiae than fasciculation, and weakness is more prominent than sensory loss in some primarily inflammatory neuropathies. Differences in axonal antigens (i.e., “structural” differences) are undoubtedly a major cause of the latter but, leaving immune specificity aside, differences in their physiological properties probably contribute: as discussed in the next section, motor axons are more prone to conduction block than sensory axons.
The same fundamental processes determine axonal excitability and impulse conduction in large myelinated axons, whether they be sensory or motor. However there are significant quantitative differences in the mechanisms that maintain axonal excitability, presumably reflecting their different activity patterns and, as a result, normal axons respond differently to insults such as ischaemia and its release and hyperventilation.7, 8 In a review in 1997, it was suggested that the expression of two depolarising conductances (/NaP and /h) was greater on sensory axons than motor, and that there was probably little difference in K+ currents or resting membrane potential.9 An unexplained difference was the extent of superexcitability in the recovery cycle,10 something that could not be accounted for by the difference in strength-duration properties.
The evidence for greater /NaP in sensory axons rested on a difference in the strength-duration properties of sensory and motor axons and on a greater effect of hyperpolarising conditioning stimuli on sensory axons in studies using “latent addition”.11, 12 The evidence for greater /h came from the finding that sensory axons accommodate more to prolonged and strong hyperpolarising currents than motor.7, 13, 14, 15
Recent studies suggest that these conclusions need to be re-interpreted.15, 16 With an updated model of the motor axon and a new model of sensory axons, Howells et al.15 examined the responses of subjects using the “Trond” protocol17, 18 that had been extended to examine the responses to hyperpolarisation in greater detail by using much stronger and much longer hyperpolarising currents than usual.14 As previously demonstrated, there were differences in strength-duration properties and in accommodation to hyperpolarising currents in the recordings from sensory and motor axons in healthy subjects (Fig. 2). However, these differences were best explained by a number of interacting differences 1) depolarisation of the resting membrane potential of sensory axons by ~4 mV, 2) a reduced slow K+ conductance on sensory axons, and 3) a shift in the voltage for half-activation of HCN channels in the depolarised direction on sensory axons, together with a difference in their expression, and 4) a greater leak conductance (which may incorporate the activity of unmodelled slow HCN isoforms) on motor axons. There was greater activity of /NaP on sensory axons, but this could be accounted for by the 4 mV depolarisation, without need to invoke greater expression of channels with persistent behaviour.
|

The model has also provided an explanation for the differences between subjects in the extent of accommodation to hyperpolarisation. This measure is perhaps the most variable measure in current excitability studies. Rather than a difference in HCN isoforms or a quantitative difference in the number of HCN channels, the differences between subjects were best explained by individual differences in the voltage for half-activation (Fig. 3).15 Accordingly the variability within a subject was found to be much less than that between subjects. This seems intuitively reasonable: healthy subjects differ in exercise and lifestyle habits, and these factors can produce differences in the gating of HCN channels. The validity of the model on which these conclusions are based has been verified recently using a totally different approach to studying axonal excitability.16 The response of human sensory and motor axons has been studied in the frequency domain using a “ZAP” (impedance [Z] amplitude profile) protocol to determine the propensity for resonant behaviour. Rather than DC conditioning stimuli, the ZAP used a small-amplitude sine wave current whose instantaneous frequency was continuously increased from start to end. Without modification, the models could explain the ZAP data recorded using a different approach to perturbing excitability. In other studies, the ability of the model to explain the different changes of sensory and motor axons during hyperthermia provides further validation of the model.19
|

An important insight from these studies is that we have been too hasty in the past to attribute changes in axonal behaviour to changes in the expression (i.e., number) of channels. Changes in the gating of channels occur commonly during normal life, e.g., through changes in membrane potential or the effects of many intracellular and extracellular modulators, and Na+ and HCN channels are particularly prone to such modulation. There is thus great potential for metabolic disturbances to affect axonal excitability, over and above any change in channel expression.
|
CLINICAL CONSEQUENCES
|
Differences in the excitability of sensory and motor axons have clinical consequences. On the one hand a more depolarised resting membrane potential and greater activity of two depolarising currents, /NaP and /h, will render sensory axons more prone to ectopic activity than motor. Paraesthesiae will develop more readily than fasciculation in response to many interventions (e.g., ischaemia, hyperventilation) or disease processes. On the other hand motor axons are less protected from hyperpolarising stresses, and therefore more prone to develop conduction block.
Axons undergo changes in membrane potential during normal activity (Table 1). For example, hyperpolarisation occurs normally when axons conduct trains of impulses.20 This is largely the result of activation of the electrogenic Na+/K+ pump to restore intra-axonal Na+ concentration, as discussed earlier. An implication of the differences between sensory and motor axons is that motor axons should hyperpolarise more in response to the same stresses. This appears to be the case. The extent of hyperpolarisation depends on the impulse load: i.e., the discharge rate and the train length,21, 22 and evidence for hyperpolarisation of human motor axons has been demonstrated during voluntary contractions as brief as 15 s (Fig. 4).23 However motor axons undergo approximately twice as much hyperpolarisation as sensory axons when they conduct impulse trains at the same physiologically meaningful frequencies (Fig. 5).24
|

|
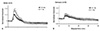
|

|
CONDUCTION FAILURE
|
The security of impulse transmission at each node of Ranvier is very high: the driving current is normally more than 5 times that required to reach threshold for initiation of the action potential. In healthy axons, the safety margin is lower at branch points (because the node must then drive the action potential in the daughter branches). Similarly fever will impair impulse conduction because it decreases the time integral of the Na+ current.19 However it is only in impaired axons with a very low safety margin that significant conduction block ensues.
When pathology results in conduction block in some but not all axons, conduction in others will be problematic.25 Normal physiological processes can then result in variations in the number of conducting fibres and the severity of the clinical deficit (Table 2), and this means that neurophysiological testing for conduction block can be improved by taking variability of the block into account (Table 3). Conduction block can occur when either the driving Na+ current is reduced, or the threshold that must be reached is elevated. The Na+ current can be reduced by heating (as mentioned above) or by axonal depolarisation. The latter occurs when the decreased availability of Na+ channels due to their inactivation outweighs the increased excitability due to the depolarisation. Similarly the Na+ current can be decreased by blocking the channels with, e.g., tetrodotoxin, as occurs in puffer fish poisoning.26
|

|

Conduction of impulse trains results in hyperpolarisation of the active axons, and this is greater in motor axons than sensory. In the presence of a severely impaired safety margin some axons may become incapable of conducting further impulses. In single human axons damaged during experiments by microneurographic electrodes, Inglis and colleagues demonstrated that activity could cause a progressive impairment of action potential generation, and ultimately conduction block (Fig. 6), with recovery of the ability to conduct after rest.27 For further discussion of the basis of the bipeaked action potentials in microneurographic studies, the reader is referred to the cited paper and Vallbo.28 Interestingly conduction seemed stable and secure if internodal conduction time was less than ~500 µs (normal value <30 µs). Above this, however, action potential generation became insecure and often failed. Also of interest, the longest internodal conduction times in axons about to undergo complete conduction block was 1.0–1.4 ms (Fig. 6). These findings are relevant when we interpret the extent of conduction slowing in nerve conduction studies.
|

In patients with multifocal motor neuropathy and chronic inflammatory demyelinating polyneuropathy, a voluntary contraction can induce or worsen the degree of conduction block.29, 30, 31 However, this phenomenon may make only a minor contribution to the clinical deficit.32, 33 Nevertheless the changes in the ability of impaired axons to conduct provide a cogent explanation for the fluctuations in deficit and fatigue that occur in demyelinating disease, particularly multiple sclerosis, where the axons are at core temperature.6
|
CONCLUSION
|
Intricate mechanisms have evolved such that axons can conduct impulse trains securely. This has resulted in differences in the fine detail of axonal function. As a consequence, motor axons are more likely to block than sensory when exposed to the same insult, and normally innocuous manoeuvres may be able to precipitate conduction block in axons critically impaired by neurological disease.
|
Acknowledgements
|
This work was supported by funding to Forefront, a collaborative research group dedicated to the study of frontotemporal dementia and motor neuron disease, from the National Health and Medical Research Council of Australia (NHMRC) program grant (#1037746), and by a Bill Gole Research Fellowship from the Motor Neurone Disease Research Institute of Australia.
|
References
|
| 1. | Vogel W, Schwarz JR. In: Waxman SG, Stys PK, Kocsis JD, editors. Voltage-clamp studies on axons: macroscopic and single-channel currents. Oxford: Oxford University Press; 1995. pp. 257-280. |
| 2. | Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1. 2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci U S A 2004;101:8168–8173. 
|
| 3. | Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. J Physiol 1987;383:45–67.
|
| 4. | Grafe P, Bostock H, Schneider U. The effects of hyperglycaemic hypoxia on rectification in rat dorsal root axons. J Physiol 1994;480:297–307.
|
| 5. | Tatsumi H, Katayama Y. Na+ dependent Ca2+ influx induced by depolarization in neurons dissociated from rat nucleus basalis. Neurosci Lett 1995;196:9–12.
|
| 6. | Vucic S, Burke D, Kiernan MC. Fatigue in multiple sclerosis: mechanisms and management. Clin Neurophysiol 2010;121:809–817.
|
| 7. | Bostock H, Burke D, Hales JP. Differences in behaviour of sensory and motor axons following release of ischaemia. Brain 1994;117:225–234.
|
| 8. | Mogyoros I, Kiernan MC, Burke D, Bostock H. Excitability changes in human sensory and motor axons during hyperventilation and ischaemia. Brain 1997;120:317–325.
|
| 9. | Burke D, Kiernan MC, Mogyoros I, Bostock H. Susceptibility to conduction block: differences in the biophysical properties of cutaneous afferents and motor axons. In: Kimura J, Kaji R, editors. Physiology of ALS and Related Disorders. Amsterdam: Elsevier; 1997. pp. 43-53. |
| 10. | Kiernan MC, Mogyoros I, Burke D. Differences in the recovery of excitability in sensory and motor axons of human median nerve. Brain 1996;119:1099–1105.
|
| 11. | Mogyoros I, Kiernan MC, Burke D. Strength-duration properties of human peripheral nerve. Brain 1996;119:439–447.
|
| 12. | Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. J Physiol 1997;498(Pt 1):277–294.
|
| 13. | Lin CS, Kuwabara S, Cappelen-Smith C, Burke D. Responses of human sensory and motor axons to the release of ischaemia and to hyperpolarizing currents. J Physiol 2002;541:1025–1039.
|
| 14. | Tomlinson S, Burke D, Hanna M, Koltzenburg M, Bostock H. In vivo assessment of HCN channel current (Ih) in human motor axons. Muscle Nerve 2010;41:247–256.
|
| 15. | Howells J, Trevillion L, Bostock H, Burke D. The voltage dependence of Ih in human myelinated axons. J Physiol 2012;590:1625–1640.
|
| 16. | Howells J, Bostock H, Burke D. Accommodation to hyperpolarization of human axons assessed in the frequency domain. J Neurophysiol 2016;116:322–335.
|
| 17. | Kiernan MC, Burke D, Andersen KV, Bostock H. Multiple measures of axonal excitability: a new approach in clinical testing. Muscle Nerve 2000;23:399–409.
|
| 18. | Kiernan MC, Lin CS, Andersen KV, Murray NM, Bostock H. Clinical evaluation of excitability measures in sensory nerve. Muscle Nerve 2001;24:883–892.
|
| 19. | Howells J, Czesnik D, Trevillion L, Burke D. Excitability and the safety margin in human axons during hyperthermia. J Physiol 2013;59:3063–3080.
|
| 20. | Bostock H, Grafe P. Activity-dependent excitability changes in normal and demyelinated rat spinal root axons. J Physiol 1985;365:239–257.
|
| 21. | Morita K, David G, Barrett JN, Barrett EF. Posttetanic hyperpolarization produced by electrogenic Na+-K+ pump in lizard axons impaled near their motor terminals. J Neurophysiol 1993;70:1874–1884.
|
| 22. | Bostock H, Bergmans J. Post-tetanic excitability changes and ectopic discharges in a human motor axon. Brain 1994;117:913–928.
|
| 23. | Vagg R, Mogyoros I, Kiernan MC, Burke D. Activity-dependent hyperpolarization of human motor axons produced by natural activity. J Physiol 1998;507:919–925.
|
| 24. | Kiernan MC, Lin CS, Burke D. Differences in activity-dependent hyperpolarization in human sensory and motor axons. J Physiol 2004;558:341–349.
|
| 25. | Park SB, Lin CS, Burke D, Kiernan MC. Activity-dependent conduction failure: molecular insights. J Peripher Nerv Syst 2011;16:159–168.
|
| 26. | Kiernan MC, Isbister GK, Lin CS, Burke D, Bostock H. Acute tetrodotoxin-induced neurotoxicity after ingestion of puffer fish. Ann Neurol 2005;57:339–348.
|
| 27. | Inglis JT, Leeper JB, Wilson LR, Gandevia SC, Burke D. The development of conduction block in single human axons following a focal nerve injury. J Physiol 1998;513:127–133.
|
| 28. | Vallbo ÅB. Prediction of propagation block on the basis of impulse shape in single unit recordings from human nerves. Acta Physiol Scand 1976;97:66–74.
|
| 29. | Kaji R, Bostock H, Kohara N, Murase N, Kimura J, Shibasaki H. Activity-dependent conduction block in multifocal motor neuropathy. Brain 2000;123:1602–1611.
|
| 30. | Cappelen-Smith C, Kuwabara S, Lin CS, Mogyoros I, Burke D. Activity-dependent hyperpolarization and conduction block in chronic inflammatory demyelinating polyneuropathy. Ann Neurol 2000;48:826–832.
|
| 31. | Nodera H, Bostock H, Izumi Y, Nakamura K, Urushihara R, Sakamoto T, et al. Activity-dependent conduction block in multifocal motor neuropathy: magnetic fatigue test. Neurology 2006;67:280–287.
|
| 32. | Straver DC, van den Berg LH, Franssen H. Activity-dependent conduction block in chronic inflammatory demyelinating polyneuropathy. J Neurol Sci 2011;300:33–38.
|
| 33. | Straver DC, van den Berg LH, van den Berg-Vos RM, Franssen H. Activity-dependent conduction block in multifocal motor neuropathy. Muscle Nerve 2011;43:31–36.
|