

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Histone deacetylases (HDACs) are the group of enzymes that remove the acetyl group from lysine residue. To date, 18 mammalian HDACs have been identified and are characterized into four classes: class I HDACs (HDACs 1, 2, 3, and 8), class II HDACs (HDACs 4, 5, 6, 7, 9, and 10), class IV (HDAC 11) and class III (sirtuin family: sirt1-sirt7). Class II HDACs are further divided into two subgroups: class IIa, which has a large C-terminus, and class IIb, which has two deacetylase domains. Class I, II, and IV HDACs need a zinc ion (Zn2+) and share a similar catalytic core for acetyl-lysine hydrolysis,1 while class III HDACs require a nicotinamide adenine dinucleotide for their enzyme activity.
Even though both class I and class II HDACs have a conserved HDAC domain, they have quite different characteristics. Class I HDACs are present ubiquitously, whereas class II HDACs are expressed in a tissue-specific manner relatively. The class I HDACs are located mainly in the nucleus, whereas class II HDACs undergo shuttling from the nucleus to the cytoplasm after phosphorylation by protein kinase C or by protein kinase D. The class IV HDAC, HDAC11 shares the catalytic domain both of class I and II HDACs. The specific role of class IV HDAC, however, still remains unclear.
Because of the Zn2+-dependent nature of the HDAC domain in class I, II, and IV HDACs, the intrinsic activity of HDACs is suppressed by inhibitors which occupy the catalytic core of the zinc-binding site.2 The specific motif of HDAC inhibitor, which fits into the tubular pocket where the Zn2+ existed originally, then interferes with the binding of the Zn2+. HDAC inhibitors can be categorized by the structure of their Zn2+-binding group: hydroxamic acids, carboxylic acid, benzamides, and cyclic peptides.3 For examples, potent non-selective HDAC inhibitors, trichostatin A (TSA) and suberanilohydroxamic acid (SAHA) are in the hydroxamic acid group, while valproate is a member of the carboxylic acid group, MS-275 is widely used benzamide group inhibitor, and Romidepsin is the representative inhibitor having a cyclic peptide structure.
HDACs have an important role both in transcription regulation and in protein modification. As HDACs remove the acetyl moiety from lysine residues at histone tails, which tighten the interaction between the positive-charged histones and the negative-charged DNA. Therefore, histone deacetylation leads to chromatin compaction and inhibits the binding of transcription machinery at the promoter region, which results in repression of mRNA synthesis. Besides the role in transcription-repression, HDACs also function as regulators in posttranslational modification (PTM). HDACs deacetylate non-histone proteins including both transcription factors, such as E2F, p53, c-Myc, and NF-κB, and signal mediators, such as Stat3, Smad7, and β-catenin which regulate cellular homeostasis.4 Since PTM determines protein activity, stability, subcellular localization, and protein-protein interactions, these diverse protein modifications by PTM modifiers are important for a proteins fate. PTM determines disease prognosis, and drugs that modulate PTM has been investigated as potentially therapeutic for several pathological disease. Imatinib mesylate successfully interrupts Abl kinase activity by blocking the tyrosine kinase domain and thereby induces apoptosis of malignant cells in chronic myeloid leukemia patients. HDACs are one of the important enzymes that regulate the PTM of other proteins, HDAC inhibitors also have therapeutic potential for refractory diseases. On the basis of the studies demonstrated that HDAC inhibition could reverse cardiovascular diseases, in the present review, we will discuss the role of HDAC and HDAC inhibitors as an important therapeutic target in cardiovascular disease beyond the anticancer properties of HDAC inhibitors.
THE ROLE OF HDACS IN TUMORIGENESIS
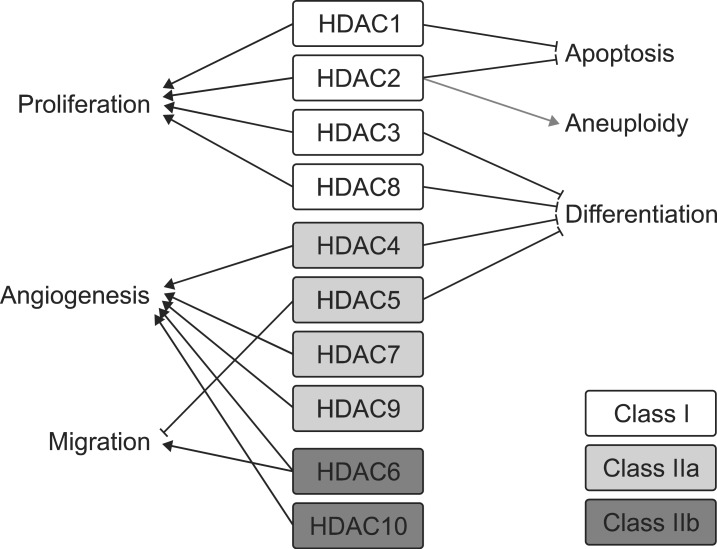
Incidentally somatic mutations allowed cells to acquire novel capacities, which benefitted their survival. After the accumulation of aberrant phenotypes in single cell in sequential events, a malignant cancer cell can be developed. According to recent studies, many HDACs are increased in malignant cells and HDACs have been closely linked with acquisition of malignant phenotypes in cancerogenesis. Class I HDACs are considered to stimulate cell proliferation and survival based on preclinical and clinical studies. For example, increased expression of HDAC1 was reported in gastrointestinal cancer,56 prostate,7 and breast8 carcinomas. Aberrant upregulation of HDAC2 has been reported in uterine, cervical,9 and gastric10 cancers. HDAC2 is regarded as the responsible factor for the loss of APC expression in colorectal carcinoma.11 Other groups have found that HDAC3 or HDAC6 was also highly overexpressed in colon and breast cancer cells, respectively.612 HDACs expression is increased in solid tumors and in hematological cancers, which correlates with a poor prognosis. The functional relevance of aberrant overexpression of HDACs is somewhat diverse among the specific subtypes. Class I HDACs generally induce cell proliferation. Furthermore both HDAC1 and HDAC2 inhibit apoptosis of cancer cells. Other HDACs, HDAC3, 4, 5, and 8, tend to inhibit differentiation and HDAC4, 6, 9, and 10 are closely linked with cancer angiogenesis while class IIb HDACs, both HDAC6 and 10, provoke cell motility, which results in metastasis (Fig. 1). Inhibitors of HDAC targeting proliferation, differentiation, angiogenesis and migration are a potential cancer therapeutic strategy.
1. Proliferative phenotype
Uncontrolled proliferation is the most distinct phenotype in cancer. The p21 gene, a potent cell-cycle arrester, seems to be correlated with HDACs. The transcription level of p21 is significantly decreased by the upregulation of HDACs in many cancer cell types. Transient overexpression of HDAC2, HDAC3, or HDAC6 induces p21-promoter silencing and subsequent down-regulation of proteins in various cell types.61314 Similarly, an increase in p21-expression and thereby reduced adenoma formation is observed either by knockdown of HDAC2 or HDAC3 by the specific siRNA or by HDAC inhibitors such as sulforaphane,13 valproate (VPA),11 and sodium butyrate.15 Another important gene for proliferation, cyclin D1, which is overexpressed in cancers, is also decreased by treatment with TSA, a non-selective HDAC inhibitor.16
2. Undifferentiated features
The cancer cells frequently remain undifferentiated status, which accords malignancy to the tumor cells. For the responsible mechanism, down-regulation of differentiation factor, GATA family, is pointed to in several cancers. Treatment with TSA in ovarian cancer cells robustly increases histone acetylation on promoters of GATA4 and GATA6.17 The restoration of both GATA4 and GATA6 results in differentiation of ovarian cancer cells. It may improve the survival rate of cancer patients because differentiated cancer cells are more susceptible to chemotherapy. HDACs have also been found to be involved in leukemogenesis. Runx1 is the definitive factor in hematopoietic linage differentiation. In the case of t(8;21), Runx1 fuses to ETO then generates the Runx1-ETO chimeric protein. Runx1-ETO (also known as AML1-ETO) chimera represses transcription both of p14ARF and c-fms by performing as a dominant negative actor against to wild-type of Runx1 which recruits HDACs for transcription-repressor complex.1819 BCL6, a BTB/POZ member which is critical for survival and differentiation, is the suppressed transcription factor both by class I and class II HDACs in B-cell lymphoma.20
3. Tumor vessel formation
Neoangiogenesis is required for both nutrient and oxygen supply in solid tumors and HDACs perform crucial roles in these processes. HDAC1 overexpression induces angiogenesis by secreting both vascular endothelial growth factors and hypoxia inducible factor-alpha. Immunohistochemical analysis reveals that HDAC1 is highly expressed in hypoxic regions of tumors whereas TSA treatment successfully inhibits hypoxia-induced neoangiogenesis, in vivo.21 Besides TSA, apicidin, and VPA are useful to prevent neoangiogenesis.2223
4. Distant metastasis
Metastasis, the spread cancer cells across tissues, occurs in many malignant tumors. Unfortunately, aberrant upregulation of HDACs allows an increase in distant metastasis of cancer cells. Forced expression of HDAC1, 6, or 8 increases cell motility in MCF-7 cells.24 E-cadherin, an important gene for cell-cell adhesion, is regulated by class I HDACs. HDAC1 and HDAC2 repress the transcription of E-cadherin correlated with Snail and mSin3A. TSA treatment abolishes Snail-mediated repression.25 HDAC3 also binds to the E-cadherin promoter after making a complex with PPARγ, and VPA depresses promoter binding of HDAC3-PPARγ which restores E-cadherin expression.26 Similarly, treatment of HDAC inhibitors using low doses suppresses local invasion of fibroblasts,27 B16-BL6 melanoma cell invasion of matrigels,28 and breast cancer cells.
5. Aneuploidy
Aneuploidy, or an abnormal number of chromosomes, is identified as a negative prognostic factor for epithelial malignancies. It is reported that aneuploidy is observed in approximately 90% of both solid tumors and hematopoietic neoplasias.29 HDAC2 was highly expressed in aneuploidy cell lines than diploid cell lines. HDAC2 nuclear immunopositive cells increased in aneuploidy colorectal cancer specimens than in diploid carcinomas.30 According to a previous study, HDAC inhibitors lead to a decrease in aneuploidy of malignant cells and an increase in euploidy. It is unclear, however, whether HDAC inhibition specifically induces apoptosis of cancer cells or induces recovery of euploidy.31
APPROVAL HDAC INHIBITORS
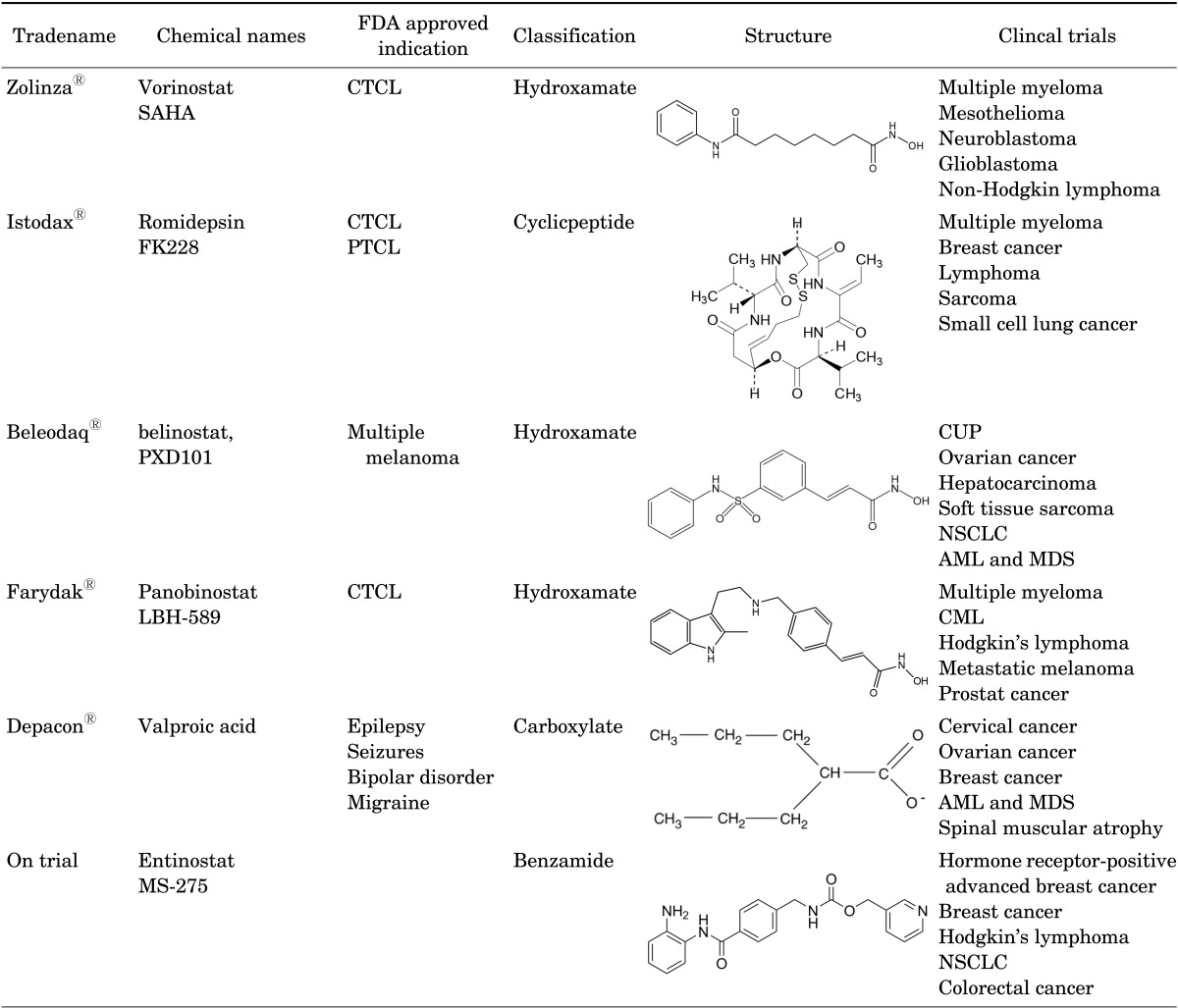
To date, the United States Food and Drug Administration (US FDA) has approved four HDAC inhibitors for the anti-cancer drugs: Vorinostat, Romidepsin, Belinostat, and Panobinostat (Table 1). Furthermore, more than five HDAC inhibitors are in clinical trial phase III including repositioning of already approved HDAC inhibitors.
1. HDAC inhibitors
Vorinostat (SAHA, trade name; Zolinza®) is a linear hydroxamate compound that was approved for the treatment of cutaneous T-cell lymphoma (CTCL) in October 2006. The objective response rate, which was determined by clinical responses, was 30%.32 Thrombocytopenia and anemia, however, were reported as severe hematologic side effects which was more frequently reported in the patients who received intravenous administration of Vorinostat.33 Several combination therapies of Vorinostat with a conventional regimen for various solid tumors are now in clinical trials up to phase III.
Romidepsin (FK228 or depsipeptide, trade name; Istodax®) is a cyclicpeptide HDAC inhibitor and was approved in November 2009. Originally the US FDA approved it for CTCL and decided to expand the indication to include peripheral T-cell lymphoma (PTCL) in November, 2011. The overall response rate was 34 % in CTCL34 and the objective response rate of PTCL was 25%.35 Additional clinical studies in solid tumors using combination therapy for pancreatic, breast, non-small cell lung cancer, and thyroid cancers are in progress.
Third FDA approval was given for the HDAC inhibitor in July 2014. Belinostat (PXD101, trade name; Beleodaq®) which is a hydroxamic acid compound licensed for the treatment of relapsed or refractory PTCL. The overall response rate was 26%.36 Like the other two FDA approved HDAC inhibitors, Belinostat is also in the clinical trial phase for solid tumors; anticancer effects against solid tumors, however, was disappointing. The effect concentration of Belinostat was not high enough because of an insufficient blood supply caused by its anti-angiogenic effects.37
The most recently approved HDAC inhibitor is Panobinostat (LBH-589, trade name; Farydak®). Panobinostat was licensed in February 2015 for the treatment of multiple myeloma. Objective responses of Panobinostat is 27%.38 Panobinostat is also a hydroxamic acid group. More cumulative data is necessary.
One carboxylate group HDAC inhibitor (VPA, as magnesium salt) and one benzamide HDAC inhibitor (Entinostat) are on phase III clinical trials. Wildly used as an anti-epileptic drug, VPA is in phase III trials for solid tumors such as cervical cancer or ovarian cancer, and even for spinal muscular atrophy. Entinostat (MS-275) is in phase III for hormone receptor-positive advanced breast cancer and is in phase II for lung cancer, breast cancer, and Hodgkin lymphoma.
2. Possible limitation of HDAC inhibitors as an anticancer drug
Apparently, HDAC inhibitors induce cell cycle arrest and thereby apoptosis in cultured cancer cells. HDAC inhibitors not only increase the transcription level of p21, a potent cell cycle arrestor, but also activate the acetylation of p53. For general effects, HDAC inhibitors stimulate transcription of proapoptotic genes such as Bax, Bak and Apaf1.39
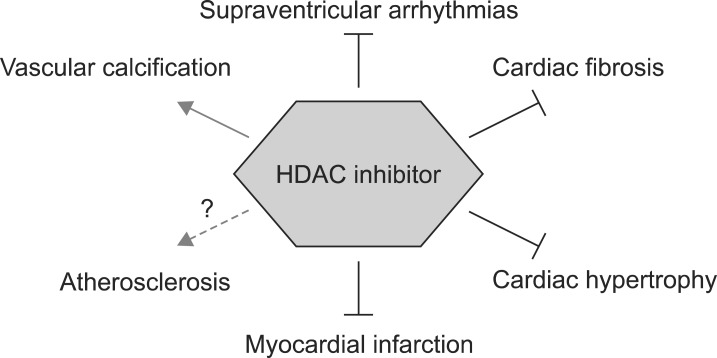
Anticancer properties in humans, however, seem to be somewhat limited. HDAC inhibitors non-specifically block angiogenesis, inflammation, and proliferation. Inhibition of tumor angiogenesis results in a failure of drug delivery to the solid tumors. Anti-inflammation activity also induces apoptosis of tumor-fighting immune cells. These unwanted side effects are regarded as possible mechanisms of the marginal effect against solid tumors. General side effects of HDAC inhibitors in chemotherapy, however, seem to be beneficial in chronic cardiovascular diseases such as atherosclerosis, myocardial infarction, arrhythmia, and transition of heart failure. A new era for HDAC inhibitors is now ready to extend to cardiovascular diseases.
THE ROLE OF HDACS IN CARDIOVASCULAR DISEASES
1. Atherosclerosis
Atherosclerosis is a chronic and a progressive disease of the arteries caused by abnormal accumulation of lipid droplets, inflammation of multifactorial cells, generation of a fibrous cap, and reactive proliferation of vascular smooth muscle cells. According to several studies, HDACs are closely linked with in the progression of atherosclerosis and HDAC inhibitors which successfully prevent the progression of atherosclerosis. Our group40 already suggested that the pan-HDAC inhibitor, scriptaid or TSA, was useful for the prevention of neointima formation from balloon injury. The transcription of p21WAF1/Cip1 was significantly increased in the HDAC inhibitor-treated group.4142 In contrast, a few groups found that HDAC inhibitors might stimulate atherogeneisis. It is noteworthy that HDAC inhibitors effectively suppress vascular smooth muscle cell proliferation, which seems to be effective for preventing atherosclerosis.
It is noteworthy that the characteristics of the certain HDAC inhibitor might result in these controversial outcomes. Global HDAC inhibitors, such as TSA, have a tendency to have a dual response: anti-inflammatory properties at low concentrations but pro-inflammatory effects at high concentrations.43 It is assumed that the role of diverse HDACs associated with single pathophysiology are quite different and the overall inhibition effect of HDAC inhibitors also somewhat varies according to its concentration. Targeted regulation of specific HDACs by their own inhibitor would be an ideal regimen.
2. Arrhythmia
Only a few reports have elucidated the therapeutic application of HDAC inhibitors in cardiac arrhythmia. TSA dramatically corrected atrioventricular conduction abnormalities in mouse hearts which were induced by a genetic disruption of HopX.44 The dramatic effect of TSA might be associated with the HopX-HDAC2 axis when considering that HopX directly recruits HDAC2.45
More direct evidence that HDACs are responsible for cardiac arrhythmia have also been suggested; myocytespecific ablation of both HDAC1 and HDAC2 results in an aberrant increase in the subunits of the calcium channel.46 Our group also detected that ion channels such as Scn3b (sodium) and Kcne1 (potassium) were dysregulated when HDAC2 was overexpressed. Dysfunction of Kcne1 may participate in Long QT syndrome and the loss of function of Scn3b is responsible for Burgada syndrome,47 which implicates that alteration of HDAC2 activity may lead to fatal cardiac arrhythmias.
3. Myocardial infarction
Ventricular myocytes are supported oxygen and nutrients through the specialized circulation system: the coronary arteries. Myocytes, however, are vulnerable to damage even by short term ischemic events because of poor anastomosis in the coronary artery. Furthermore, the myocytes received alternate damage after revascularization. This serial injury is termed ischemia-reperfusion (I/R) injury.
HDAC inhibitors have been highlighted as promising drugs to reduce I/R injury, to ameliorate cardiac dysfunction, and to minimize infarction size.484950 An ex vivo study by use of the Langendorff system reveals that preconditioning of TSA preserves cardiac performance after I/R injury. Preconditioning by injection of TSA before the I/R injury reduces the infarction area and restores contractile dysfunction.48 Furthermore, HDAC inhibitors improve fatty acid oxidation by restoring PGC-1α in I/R injuries.51 To date, it is regarded that the major advantageous effects of HDAC inhibitors in I/R injury is mediated by inhibition of generation of immature vasculatures, by reducing inflammation, or by facilitation of energy metabolism.
HDAC inhibitors are also beneficial for minimizing the scar size of myocardial infarction (MI). The infarction area generated by permanent ligation of the left anterior descending artery is dramatically reduced by administration of HDAC inhibitors such as tributyrin, VPA, or TSA.495052 It has also been reported that administration of TSA for 2 months markedly prevented cardiac dysfunction and suppressed cardiac remodeling.52 Despite some contradictory reports about HDAC inhibitors in acute coronary syndromes, it is more commonly shown that HDAC inhibitors are effective both for preventing cardiac dysfunction and cardiac remodeling after MI.
4. Cardiac hypertrophy
Cardiac hypertrophy is a kind of adaptation to the increased hemodynamic demand from peripheral tissue or from another underlying diseases such as hypertension, valvular dysfunction, and MI.53 The initial adaption might be physiologic, however, cardiac hypertrophy is the beginning of the global remodeling of the heart. The roles of the HDACs in cardiac hypertrophy are being widely studied by a number of research groups including ours.45465455565758596061626364 Both classes of HDACs, class I and class IIa, are associated with the development of cardiac hypertrophy, however, they perform definitely opposite roles. Genetic ablation of HDAC2 results in resistance to various hypertrophic stimuli.64 Heart-specific overexpression of HDAC2 itself induces cardiac hypertrophy.5564 Although HDAC2 clearly provokes cardiac hypertrophy, the protein levels of HDAC2 are not altered during the process. The intrinsic activity of HDAC2 is increased in response to hypertrophic stimuli by the activated-CK2α1.5556 As for class I HDACs, there has been no clear evidence of class I HDACs other than HDAC2 in cardiac hypertrophy found. Only HDAC3, however, might allow for a transient proliferative potential to cardiomyocyte in the perinatal period.65
By contrast, class IIa HDACs negatively regulates cardiac hypertrophy. Global deletion of HDAC962 or HDAC559 shows an exaggeration of hypertrophic phenotypes. In basal conditions, class IIa HDACs capture MEF2 and interfere with the binding to its motif which results in the suppression of the transcription activity of MEF2. Class IIa HDACs are recognized by a shuttling molecule named 14-3-3 after phosphorylation by PKC/PKD or CaMKII and undergo shuttling out from the nucleus to the cytoplasm. The redistribution of class IIa HDACs causes reactivation of arrested-fetal gene programs which are regulated by MEF2, resulting in cardiac hypertrophy.596166
Because those two classes of HDACs perform opposite functions, the overall efficacy of global HDAC inhibitors in cardiac hypertrophy is questioned. We58 and other research groups6063 have suggested that cardiac hypertrophy can be completely abolished either by non-specific HDAC inhibitors545860 or even by selective class I HDAC inhibitors.586367 To summarize this phenomenon, the anti-hypertrophic properties of the non-selective HDAC inhibitor are mediated by specific regulation of class I HDACs. In addition, recently our group suggested crosstalk between HDAC2 and class IIa HDACs in the development of cardiac hypertrophy. Acetylation of HDAC2 preceded phosphorylation and those modifications were mandatory for activation of HDAC2. HDAC5, a class IIa HDAC, functioned as an enzyme that regulated acetylation of HDAC2. HDAC2 was one of the important pro-hypertrophic mediators regulated by class IIa HDACs.68
A quite recent report clearly demonstrated the role of HDAC3 in high blood pressure and the therapeutic applications of HDAC inhibitors for hypertension control.69 One more report suggest that HDAC4 induces hypertension through vascular inflammation and TSA treatment dramatically ameliorates high blood pressure.70 Taken together, this data suggests that HDAC is a novel therapeutic target for regulation of hypertension.
5. Cardiac fibrosis
Cardiac fibrosis associated with hypertrophy is notable in cardiac disease. Cardiac fibrosis results in a loss of elasticity and in insufficient dilation of the contractile chamber in the diastole phase, which is regarded as the major pathophysiology of heart failure with preserved ejection fractions (HFpEF).71 HDAC inhibitors also dramatically blocks cardiac fibrosis.555860 Fibrosis is directly inhibited by HDAC inhibitors rather than secondary changes after improving cardiac hypertrophy. More evidence enforces the postulation that HDAC inhibitors directly regulate transdifferentiation of fibroblasts to myofibroblasts.72 During scar formation, the HDAC2 protein amount is dramatically increased73 and renal fibrosis is also successfully controlled by HDAC inhibitors.7475
Very recently, the European society of cardiology and the American heart association alert the severity of HFpEF, respectively. They summarized the clinical outcomes of HFpEF patients during the last two decades who got the conventional regimen for heart failure with reduced ejection fraction (HFrEF). Strikingly, the standard strategy for HFrEF such as beta-blockers, angiotensin converting enzyme inhibitors/angiotensin receptor blockers, or aldosterone-antagonists did not sufficiently reduce the disease progression of HFpEF. According to rodent HFpEF, however, HDAC inhibition is the most promising and reproducible strategy to reverse and prevent HFpEF to normal heart. Therefore, HDAC inhibitors should be considered to start clinical trials for HFpEF.71
6. Angiogenesis
Besides general anti-neoplasmic potential, HDAC inhibitors also block angiogenesis. For example, it is well known that Vorinostat and TSA interfere with the sprouting of capillaries from aorta76 Even though the diverse physiologic activity of HDACs, it is generally accepted that class II HDACs such as HDAC4,77 HDAC5,78 HDAC6,7980 and HDAC78182 are pro-angiogenic.
In contrast with the general concept that HDAC inhibitors inhibit angiogenesis, several studies reported positive outcomes correlated with neoangiogenesis. Long-term treatments of VPA in cerebral infarction resulted in enhancement of neovascularization, reduction of infarction size, and alleviation of cerebral functions.83 Including these reports, many studies suggested that long-term treatment with HDAC inhibitors induces neovascularization.5284
In summary, the global effects of HDAC inhibitors in angiogenesis seem to be controversial. According to recent data, HDAC inhibitor preferentially regulates immature vascularization such as tumor vessels or acute ischemic sprouting.85 Long-term administration of HDAC inhibitors promote intact angiogenesis rather than weak and leaking vessels. More specific studies would be necessary to solve these problems.
7. Vascular calcification
It should be considered that in spite of the general benefits of HDAC inhibitor in the cardiovascular diseases, HDAC inhibition may cause harmful results at least in some conditions. For example, HDAC inhibition seems to aggravate the progression of vascular calcification. TSA86 and apicidin (Kwon et al., unpublished data) accelerated the calcification in vitro. E3 ligase MDM2 was dramatically increased in calcification-provocation stimuli, which induced polyubiquitination and subsequent degradation of HDAC1. The loss of activity of HDAC1 plays a crucial role in the progression of vascular calcification. Hence, HDAC inhibitors that reduce HDAC activity in vivo may accelerate vascular calcification. Considering that one of major side effect of Vorinostat is vascular calcification, HDAC inhibitor should be carefully administrated to patients who suffer from atherosclerosis or have a proatherogenic condition such as chronic renal failure or diabetes mellitus.
MISCELLANEOUS DISEASES
Besides cancer treatment, HDACs has been studied in various non-cancer diseases including neurodegenerative disease, inflammatory disease, and osteoporosis beyond cardiovascular diseases. For example, a Danish group has tested HDAC inhibitors as an HIV treatment.87 In neurological diseases, HDAC inhibitors improve the performance of learning and memory,88 Alzheimer disease,89 ischemic stroke,90 Huntington's disease.91
Several studies have implied that HDAC inhibitors may improve learning ability and memory formation. The brain slices which were acquired from Vorinostat-treated rat hippocampus showed increased synaptic functions ex vivo.9293 Moreover, RGFP966, a class I HDAC inhibitor, treated rats tended to be faster than the vehicle group in learning and in acute memory formation.94 More specifically, the synapse number of HDAC2 overexpression mice was significantly decreased. The learning impairment of HDAC2 overexpression mice was alleviated by chronic treatment with Vorinostat.95
HDAC inhibitors also might be noteworthy for testing the therapeutic potential of bone diseases such as osteoporosis and fractures. Both the protein stability and transcription level of the master regulator of bone formation, RUNX2, are specifically regulated by HDAC inhibitors.96 Furthermore, HDAC inhibitors suppress osteoarthritis using their anti-inflammation effects on joint inflammation and thereby cartilage degeneration.97
CONCLUSION AND FUTURE PERSPECTIVE
In this review, we summarize 1) the roles of HDAC in tumorigenesis and the current uses of HDAC inhibitors in cancer, 2) the functional relevance of HDACs and the therapeutic potentials of HDAC inhibitors in cardiovascular diseases (Fig. 2). According to clinical and nonclinical studies, HDAC inhibitors might result in cell-cycle arrest, apoptosis, differentiation, anti-inflammation, anti-invasion, and anti-proliferation. In short, HDAC inhibitors have general anti-tumor activities. Four HDAC inhibitor approved by the US FDA are beneficial to hematological malignancy but are having disappointing results in solid tumors as a single drug therapy. The limited effects on solid tumors seem to be a failure of the drug delivery into the cancer tissue because of insufficient blood supply caused by its anti-angiogenetic effect.37
HDAC inhibitors are also beneficial for preventing the disease progression of several cardiovascular diseases. According to studies using various HDAC inhibitors, several cardiovascular diseases would be benefit from novel indications for HDAC inhibitors including cardiac arrhythmia, myocardial infarction, global cardiac remodeling, HFpEF including hypertrophy, hypertension, and cardiac fibrosis. However, they may worsen vascular calcifications. Actually, thrombus is reported as a serious and quite frequent side effect of Vorinostat in patients.
To date, a great number of HDAC inhibitors are in clinical trials. Four drugs, Vorinostat, Romidepsin, Belinostat, and Panobinostat are already approved by the U.S. FDA for CTCL, PTCL, and multiple melanoma. Entinostat and VPA are in phase III trials for solid tumors. MGCD0103, PCI-24781, SB939, 4SC-201, ITF2357, CI-994, SRT501, and JNJ-26481585 are in phase II trials.
HDAC inhibitors represent a novel, promising regimen in various refractory diseases. Ubiquitous expression of various HDAC isoforms, however, should be noted when HDAC inhibitors are developed for certain diseases. Ideal HDAC inhibitors for cancer treatment must not block good HDACs but inhibit bad HDACs. In some case, HDAC10 is closely linked with suppression of cervical cancer metastasis.98 Hence, novel approaches should be considered to regulate bad HDACs specifically. We already reported that the inhibition of the HDAC2-activating enzyme, CK2α1, is as effective as an HDAC inhibitor in the development of cardiac hypertrophy. When regarding that HDACs generally regulated by post-translational modification novel therapeutics that target HDAC-regulating enzymes would be more ideal drugs. Novel therapeutics would be promising with the understanding of the certain PTM enzymes that regulate the specific HDACs.
 XML Download
XML Download