

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cross-talk between metabolic organs can be utilized as a means for identification of the complex pathology of chronic inflammatory diseases [1]. Adipose tissue has a dynamic and important role in the development of several metabolic-related diseases, including type 2 diabetes, atherosclerosis, and coronary heart disease (CHD) [2,3].
In humans as well as in other animal species, adipose tissue is the main site for triglyceride and cholesterol storage [4]. Studies have shown that excessive cholesterol accumulation can be toxic to several tissues, including heart, aorta, and liver [1,2,5,6]. However, scant attention has been devoted to the effects of excessive cholesterol accumulation in adipose tissue. Recent studies in LDL receptor knock-out mice (LDLR -/-) have shown that excess cholesterol accumulation in epididymal adipose tissue induces insulin resistance, impairs adipocyte differentiation and maturation, induces secretion of pro-inflammatory cytokines, and stimulates macrophage (Mø) recruitment [2]. In vitro studies using 3T3 cells have shown that cholesterol overload in adipocytes can induce endoplasmic reticulum (ER) stress and trigger the unfolded protein response, leading to activation of both c-Jun N-terminal kinases and IκB kinase, proteins that are mediators of inflammatory cytokine production. Release of cytokines, such as tumor necrosis factor (TNF)-α, results in infiltration of neutrophils, Mø, T-cells, and other immune cells [7]. In addition to these effects, cholesterol accumulation in adipose-derived stromal cells leads to impairment of adipocyte development and function [8].
Dietary interventions using low carbohydrate (L-CHO) diets have been shown to reduce serum triglycerides (TG) and increase HDL cholesterol (HDL-C).They also promote weight loss and exert beneficial effects on several chronic inflammatory diseases, including atherosclerosis, diabetes, hypertension, and dyslipidemia [9,10,11,12,13].
Guinea pigs have several similarities to humans in regard to lipoprotein and cholesterol metabolism and have been shown to be a good model for study of diet-induced atherosclerosis [10,11,14]. In this study, we sought 1) to examine the effects of a dietary cholesterol challenge on characteristics of adipose tissue, aortas, and hearts of guinea pigs and 2) to determine the extent to which low (L-CHO) or high carbohydrate (H-CHO) diets could reverse detrimental effects generated by excessive cholesterol intake. We hypothesized that high cholesterol would induce cholesterol accumulation, inflammation, macrophage infiltration, and atherosclerosis in adipose tissue and that L-CHO diets would be more efficient than H-CHO diets in attenuating these deleterious effects.
MATERIALS AND METHODS
Animals and Diets
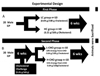
Guinea pigs weighing between 200-250 g were housed in pairs in a metal cage in a light cycle room (light from 07:00-19:00 h) with free access to water. Food consumption was monitored every other day, and guinea pigs were weighed weekly to ensure appropriate food consumption. In the first experiment, 20 male guinea pigs were randomly assigned to one of two dietary treatments: low cholesterol (LC) (0.04 g/100 g) (n = 10) or high cholesterol (HC) (0.25 g/100 g) (n = 10) for six weeks. This amount of cholesterol is equivalent to 1800 mg/d for humans and it has been shown to induce hepatic cholesterol accumulation and hypercholesterolemia in guinea pigs [10,11]. In the second experiment, 20 guinea pigs were fed HC for six weeks and then randomly assigned to one of two dietary treatments for an additional six weeks, either a L-CHO diet containing 0.04 g/100 g of cholesterol and the following energy distribution: 60% fat, 10% CHO and 30% protein, or a H-CHO diet with the same concentration of dietary cholesterol and the following energy distribution: 30% fat, 55% CHO, and 15% protein diet. Vitamins and minerals were adjusted to meet requirements for guinea pigs [10]. Details on composition of the diets are shown in Table 1. Diets were prepared and pelleted by Research Diets Inc. (New Brunswick, NJ). At the end of the 12 weeks, guinea pigs were sacrificed after a 12-hour fast. Blood, aortas, hearts, and epididymal adipose tissue were harvested and stored at -80℃ for further analysis. Diet assignment is illustrated in Fig. 1. Animal experiments were performed in accordance with U.S. Public Health Service/U.S. Department of Agriculture guidelines. Experimental protocols were approved by the IACUC at the University of Connecticut. Protocol number was A11-023.
Fasting plasma lipids
Plasma HDL-C, total cholesterol (TC), LDL-C, and TG were analyzed using the Cobas c111 analyzer (Roche Diagnostics, Indianapolis, IN). This instrument uses direct enzymatic methods with photometric detection for calculation of plasma lipid concentrations (Roche Diagnostics, Indianapolis, IN).
Cholesterol accumulation in aorta and adipose tissue
Aortic (~0.5 g) and adipose tissue (~1 g) aliquots were homogenized and added to 15 ml of chloroform-methanol 2:1 (v/v). These aliquots were extracted with an additional 10 mL of the same solution and mixed. The extracts were filtered through Whatman paper no.1 for removal of denatured protein. TC and free cholesterol (FC) were measured using enzymatic kits (Wako Chemicals USA, Inc., Richmond, VA] according to Carr et al. [15]. The esterified cholesterol fraction was calculated by subtracting free from total cholesterol [15].
Adipose inflammatory cytokine concentrations
Epididymal adipose tissue was homogenized for measurement of concentrations of cytokines TNF-α, Interleukin (IL)-10, IL-2, IL-10, monocyte chemotactic protein-1 (MCP-1), and granulocyte macrophage colony-stimulating factor (GM-CSF) using Luminex technology (Luminex 200 System, Austin, TX) with the MILLIPLEX MAP Mouse Cytokine Immunoassay kit (Millipore corporation, Charles, MO, USA), as previously described [16].
Arterial morphology
For atherosclerosis evaluation, hearts were immersed in formalin, and paraffin sections of 3-5 µm were sliced for preparation. The slides were stained with hematoxylin and eosin and evaluated via light microscopy as previously reported [14].
Macrophage accumulation in adipose tissue
Single-label immunohistochemistry was performed in the adipose tissue. Methodology is described in detail elsewhere [16]. Macrophages were detected using a mouse monoclonal antibody Mac 387 designed for guinea pigs. In addition, cross sectional areas of adipocytes were measured using image analysis software (Image Pro; Media Cybernetics).
Statistical analysis
A non-paired Student's t test was used to evaluate effects of cholesterol challenge on heart and adipose lipids, inflammatory cytokines, and number of macrophages per mm2 between the LC and HC groups. Similarly, a non-paired Student's t test was used to evaluate the same parameters in comparing the L-CHO with the H-CHO group. P < 0.05 was considered significant.
RESULTS
Comparisons between HC and LC groups
Fasting plasma lipids, Body Weight and Organ Weight
Guinea pigs fed the HC diet gained less weight at the end of the dietary treatment (Table 2), which was associated with lower food intake (Table 2). Higher plasma LDL cholesterol was observed in guinea pigs fed the HC compared to those fed the LC diets (P < 0.0001) (Table 2). There were no differences in plasma TG or HDL-C (P > 0.05) (Table 2).
Adipose and aorta cholesterol concentration
No significant differences in epididymal adipose tissue weight were observed between the HC and the LC groups (Table 2). Despite no differences in aorta total or free cholesterol concentration between the LC and HC groups, the HC group showed significantly greater accumulation of esterified cholesterol, a known feature of early atherosclerosis (Table 3). However, there was no presence of atherosclerotic lesions in either the LC or HC groups, indicating that six weeks was not enough time for development of atherosclerosis in guinea pigs (data not shown).
Cytokine Evaluation
Higher protein concentrations of pro-inflammatory cytokines TNF-α, MCP-1, and GM-CSF were observed in adipose tissue of guinea pigs in the HC group, when compared with the LC group (Table 3). Accordingly, lower levels of the anti-inflammatory cytokine IL-10 were observed in the HC group.
Adipose Evaluation
Macrophage detection with mac387 revealed remarkably greater infiltration in adipose tissue in the HC group than in LC-fed guinea pigs (Fig. 2, panel A). Guinea pigs fed the HC diet had three times more macrophage infiltration than those in the LC group. Of particular interest, changes in adipocyte morphology were observed between LC and HC groups (Fig. 2, panel B). Adipocyte size was smaller in the cholesterol challenge animals compared with the LC group (2155 ± 229 µm2 vs 1090 ± 123 µm2, P < 0.05) (Fig. 2, panel C)
Comparisons between L-CHO and H-CHO Groups
There were no significant differences in body weight; 707 ± 134 g vs 748 ± 91 g for L-CHO and H-CHO groups, respectively (Table 4). However, and possibly associated with the high caloric content in the LC diet, guinea pigs fed LC consumed 20.5 ± 1.0 g/d compared to 25.5 ± 0.8 g/d consumed by guinea pigs fed the LC diet (P < 0.01) (Table 4).
Fasting Plasma lipids
No significant differences in plasma total LDL-C or TG were observed between the L-CHO and the H-CHO groups. The only significant difference was that higher HDL-C was observed in the L-CHO group. Values were: 0.77 ± 0.15 vs 0.39 ± 0.08 mmo/L (P < 0.05) (Table 4).
Adipose and Aorta Cholesterol Concentrations
When compared with the L-CHO guinea pigs, the H-CHO group showed higher TC concentrations in the adipose tissue and higher FC and TC concentrations in the aorta (Table 5) while the CE was lower in the L-CHO group (P < 0.05).
Cytokine Evaluation
After the cholesterol challenge, animals following the L-CHO diet showed lower concentration of pro-inflammatory cytokines TNF-α, MCP-1, and IL-2 in adipose tissue (P < 0.05) when compared with the H-CHO group (Table 5).
Adipose evaluation
We observed increased macrophage infiltration indicated by the amount of macrophages detected with the mac387 antibody in guinea pigs following the H-CHO vs. animals following the L-CHO (Fig. 3, panel A). Concentration of macrophages was 2.29-fold higher in the H-CHO group than in the L-CHO group (Fig. 3, panel B). The adipocyte size was also lower in the H-CHO group when compared to guinea pigs following the L-CHO diet (1857 ± 217 µm2 vs 2857 ± 257 µm2 (P < 0.05). (Fig. 3, panel C).
DISCUSSION
Guinea pigs are a well-established model for study of chronic inflammatory diseases. Previous studies have shown that the use of a cholesterol challenge can induce atherosclerosis, and hepatic cholesterol accumulation [10,17,18]. The aim of this study was to determine the effects of cholesterol accumulation in adipose tissue and to evaluate the metabolic effects of L-CHO diets in this animal model after development of cholesterol-induced inflammation. We found that dietary cholesterol challenge in guinea pigs triggers adipose tissue inflammation and dysfunction. In addition, we observed that the use of a low carbohydrate diet reduced adipose inflammation and promoted development of larger, more responsive adipocytes.
Cholesterol accumulation differentially impacts various cell types. In hepatocytes, cholesterol accumulation leads to depletion of mitochondria antioxidants such as glutathione, which sensitizes hepatocytes to TNF signaling, leading to increased apoptosis and accelerated nonalcoholic steatohepatitis (NASH) [5]. Several other consequences of cholesterol accumulation in the plasma membrane have been observed in the brain and other cell types such as immune cells [8,19]. Cholesterol challenge diets have been previously shown to increase levels of circulating oxidized LDL (Ox-LDL) [12,17]. This pro-atherogenic lipoprotein is internalized by scavenger receptor B1 (SR-B1) and LDL related protein 1 (LRP-1) in Mø and smooth muscle cells leading to cell dysfunction and release of pro-inflammatory cytokines and chemo-attractants and finally the formation of foam cells, the hallmark of atherosclerosis [20]. Since adipocytes express high levels of LRP-1 and SR-B1, it is likely that ox-LDL is avidly taken up by these cells leading to effects similar to those found in vascular smooth muscle cells (VSMC) and Mø [3,21]. In this study we observed that cholesterol accumulation in adipose tissue increased macrophage infiltration and was followed by elevated levels of inflammatory cytokines TNF-α, MCP-1, and GMF-CSF. There was also a significant reduction in the anti-inflammatory cytokine, IL-10 in adipose tissue from HC-fed guinea pigs. In addition, we observed less developed adipocytes, indicated by the decreased size compared with the LC group. These observations are in accordance with findings of a recent study using mouse adipose-derived stromal cells where it was observed that cholesterol accumulation led to decreased gene and protein expression of sterol regulatory binding-element protein 1 (SREBP-1) and peroxisome proliferator-activated receptor γ2 (PPAR-γ2), resulting in adipocyte dysfunction and blunted cell development [8]. Expression of SREBP-1 has been shown to directly modulate the expression of the PPARγ gene at the transcription level [22,23]. PPARγ is directly involved in the transcription of several lipogenic genes that are crucial in adipocyte development [24,25]. Dysfunctional adipocytes have been shown to be more receptive to TNF-α signaling and subsequent apoptosis, which may lead to macrophage infiltration and secretion of inflammatory cytokines, which can then result in a pro-inflammatory feedback loop [2]. Studies using LDL-/- mice reported similar results, where a dietary cholesterol challenge along with a diabetogenic diet induced increased macrophage infiltration in adipose and aortic tissue leading to development of atherosclerosis along with increased levels of systemic inflammation markers serum amyloid A and C-reactive protein [2]. Thus, our results suggest that a dietary cholesterol challenge also leads to arrest of adipocyte development and induces macrophage infiltration and inflammation in adipose tissue in guinea pigs, which allows for testing dietary interventions that can reverse these conditions.
Reverse cholesterol transport is a complex mechanism where HDL brings cholesterol from peripheral tissues to the liver to be secreted in the form of bile [26]. L-CHO diets have been shown to increase levels of circulating HDL-C [10,11,12,18]. Cholesterol in adipose tissue is actively removed by nascent and mature HDL by different mechanisms involving ABCA-1 and SR-B1 receptors [27]. According to recent studies in LDL-/- mice, cholesterol exchange in adipose tissue accounts for at least 15% of the HDL-cholesterol pool [28]. In this study we observed decreased levels of cholesterol accumulation in cholesterol-challenged animals following a L-CHO diet when compared to the challenged group, which followed a H-CHO diet. These results could in part be attributed to the increased HDL-C concentrations observed in the L-CHO group. In addition, guinea pigs following L-CHO diets showed decreased levels of inflammatory cytokines MCP-1, TNF-α, and IL-2, decreased macrophage infiltration, and increased adipocyte size. As discussed above, the increased cholesterol accumulation triggers adipocyte dysfunction, which develops into an adipose tissue pro-inflammatory state characterized by macrophage infiltration and higher pro-inflammatory cytokine levels as seen in the H-CHO group. In addition, previous studies have demonstrated that H-CHO diets increase levels of ox-LDL [10,11,12]. Another possibility on how H-CHO induces inflammation in this tissue relies on the association of circulating glucose with several proteins and lipoproteins. This association can result in irreversible advanced glycated end-products (AGEs). These products actively associate with circulating LDL changing the structure and facilitating the oxidation of this lipoprotein [29]. Adipocytes and macrophages within adipose tissue can actively uptake ox-LDL since SR-B1 is ubiquitously expressed in both cells [27]. Several studies have proven the pro-inflammatory effects of ox-LDL tissue accumulation [30,31,32] and this is likely to be a contributing factor in the increased adipose tissue inflammation observed in H-CHO animals when compared with the L-CHO group. We also observed that adipose cholesterol was decreased in the L-CHO group.
Finally, another factor contributing to the cholesterol decrease seen in adipose tissue of the L-CHO group is that guinea pigs were probably in a state of ketosis due to the very minimum levels of CHO (10% of energy) in their diet. Thus,it is possible that a greater portion of acetyl-CoA derived from fat oxidation was diverted to formation of ketone bodies as opposed to cytoplasmic cholesterol synthesis [34].
The results of this study confirm that excessive dietary cholesterol induces cholesterol accumulation in adipose tissue, promotes adipose tissue inflammation, and increases macrophage recruitment. We also observed that HC increases esterified cholesterol in aorta, which is a known marker for atherosclerotic lesion formation. In addition, we found that L-CHO diets are more effective than H-CHO diets in attenuating these detrimental effects, which confirms beneficial effects reported for humans (Reviewed in 18). This study provides insight into the role of tissue cholesterol accumulation in triggering diseases such as type 2 diabetes and CHD, generated through chronic low grade systemic inflammation. In addition, this study helps to clarify the effect of macrophage infiltration in adipose tissue on systemic low grade inflammation. Given these findings, L-CHO diets may be a therapeutic option against vascular and adipose tissue dysfunction accompanied by systemic low grade inflammation.







 XML Download
XML Download