

 PDF
PDF ePub
ePub Citation
Citation Print
Print



Introduction
The etiology of breast cancer is related to multiple factors, a significant one of which is estrogen exposure (Colditz, 1998; Grodstein et al., 1997; Service, 1998). 4-Hydroxyestradiol (4-OHE2) is hydroxylated metabolite of 17β-estradiol formed by CYP1B1. This catechol estradiol is further oxidized by sequential one-electron oxidations to produce the estrogen semiquinone radical and estrogen quinone. In rodent models, estrogens have been shown to act as carcinogens through a mechanism that involves oxidative stress in various tissues including kidney, liver, and breast (Han & Liehr, 1994; Harvell et al., 2000; Yager & Liehr, 1996).
Several cellular defense molecules including superoxide dismutase (SOD) and reduced glutathione (GSH) protect cells from oxidative damage. Manganese superoxide dismutase (MnSOD) catalyzes the conversion of superoxide to hydrogen peroxide in mitochondria. It has been demonstrated that MnSOD levels are lowered in certain cancer cells (Hitchler et al., 2006), while other studies have shown that the stimulated expression of MnSOD suppresses malignant phenotypes in mammalian tumor cells, including breast (Li et al., 1998), prostate (Liu et al., 1997), and oral carcinoma cells (Liu et al., 1998). The GSH/GSSG ratio is tightly regulated under normal circumstances, while the balance shifts towards a low GSH/GSSG ratio under conditions of oxidative stress. The ratio of GSH/GSSG measured in the blood of patients with colon and breast cancer has been found to be significantly decreased compared to that of control subjects (Pastore et al., 2003).
Several proteins are involved in oxidative DNA damage repair, and therefore genetic defects in these proteins increase the risk of developing breast cancer. Breast cancer susceptibility gene 1 (BRCA1), for example, regulates cell cycle progression and DNA damage repair, and maintains genomic integrity (Rosen et al., 2003). In addition, BRCA1 stimulates antioxidant response element-driven transcriptional activity and shifts the cellular redox balance to a higher ratio of reduced to oxidized glutathione (Bae et al., 2004). Inherited mutations or deficiencies in BRCA1 predispose its carriers to higher risks of breast and ovarian cancers (King et al., 2003; Walsh et al., 2007). Poly (ADP-ribose) polymerase-1 (PARP-1), a nuclear member of the PARP superfamily, binds rapidly to DNA single and double strand breaks where its activation facilitates DNA repair (Ame et al., 2004). In addition, PARP-1 is essential for the regulation of BRCA1 in response to DNA damage (Tong et al., 2007). PARP-1 complementation partly rescues defective DNA damage response resulting from defective BRCA1. Hence, BRCA1 and PARP-1 are thought to respond to cellular oxidative stress including exposure to the oxidized estradiol metabolites.
Cancer patients have been shown to have low concentrations of tissue and plasma tocopherols compared to their healthy counterparts (Abiaka et al., 2001; Goodman et al., 2003; McMillan et al., 2002). The functions of tocopherols in scavenging lipid peroxyl radicals in biological membranes are well documented (Baba et al., 2005). A recent report has demonstrated that tocopherols can also directly suppress ROS production by interfering with the assembly of membrane-bound NADPH-oxidase complexes (Wittmann et al., 2005). Tocopherols also regulate the expression of genes related to cell proliferation, cell cycle progression, and apoptosis in an antioxidant-independent way (Campbell et al., 2003; Campbell et al., 2006; Gysin et al., 2002; Roy et al., 2002).
In this study, we hypothesized that continuous exposure of normal breast epithelial cells to 4-OHE2 increases oxidative stress and disrupts redox balance thereby altering the availability of repair elements. Effects of α- or γ-tocopherol on the markers of oxidative stress and the expression of DNA repair elements were also examined.
Materials and Methods
Chemicals
All chemicals were purchased from Sigma-Aldrich (St. Louis, MO, USA) unless stated otherwise. Cholera toxin was obtained from List Laboratories (Campbell, CA, USA). Culture media, epidermal growth factor, penicillin-streptomycin, horse serum and glutamine were purchased from Invitrogen (Carlsbad, CA, USA). Anti-MnSOD, BRCA1, and HRP-conjugated secondary antibody were obtained from Upstate (Lake Placid, NY, USA). The glutathione assay kit was obtained from Cayman Chemical (Ann Arbor, MI, USA). PRO-PREP™ was purchased from iNtRON Biotechnology (Daejeon, Korea). The Bradford protein assay kit was purchased from BioRad laboratories (Hercules, CA, USA).
Cell culture
Human mammary epithelial cells (MCF-10A) obtained from ATCC (Manassas, VA, USA) were cultured in DMEM/F12 containing 10 µg/ml bovine insulin, 100 ng/ml cholera toxin, 0.5 µg/ml hydrocortisone, 20 ng/ml recombinant human epidermal growth factor, 2 mM L-glutamine, 100 µg/ml penicillin/streptomycin, and 5% horse serum at 37℃ in an atmosphere of 5% CO2. The non-tumorigenic MCF-10A cell line was originally derived from a patient with proliferative breast disease (Cuendet & Bolton, 2006).
Cell treatment
MCF-10A cells were incubated with 0.1 µM 4-OHE2, either with or without 30 µM tocopherol for 24 to 96 h. 4-OHE2 and tocopherols were replaced every day during the incubation period.
Measurement of intracellular ROS accumulation
MCF-10A cells were washed and treated for 20 min with 100 µM 2', 7'-dichlorodihydrofluorescein diacetate (DCFH-DA), a nonfluoroscent probe that diffuses into cells. Cytoplasmic esterase hydrolyzes DCFH-DA to 2', 7'-dichlorodihydrofluorescein (DCFH) and then ROS generated within cells oxidize DCFH to the fluorescent dye, 2', 7'-dichlorofluorescein (DCF). The intensity of DCF was measured by a flow cytometry (FACS Calibur, Becton Dickinson, USA) at 530 nm using an FL1 filter.
Measurement of GSH/GSSG ratios
MCF-10A cells were collected by centrifugation at 1000×g for 5 min at 4℃. The cell pellet was sonicated in 1 mL PBS (pH 7.2) and centrifuged at 13,000×g for 20 min. Total GSH and GSSG were measured using a glutathione assay kit. The GSH disulfide (GSSG) content of cells was determined by adding 10 µL 2-vinylpyridine solution per mL of sample in order to remove GSH according to the method described by Griffith (1980).
Protein extraction and western blot analysis
Cells were harvested by trypsinization, washed with PBS (pH 7.2) and the pellets were collected. The pellets were extracted using the PRO-PREP™ and protein concentration was determined. Protein (30 µg) was added to each well of 10-12% SDS-PAGE. The gel was run at 100 V, 250 mA and transferred to a PVDF membrane (Amersham Pharmacia Biotech, Piscataway, NJ, USA) for 80 min. The membrane was blocked in blocking buffer (0.2% Tween 20 in PBS, pH 7.2), containing 5% non-fat dry milk for 2 h at room temperature. Dilution of primary antibodies (MnSOD 1:2000, BRCA1 1:250, PARP-1 1:500) was made in blocking buffer. The blots were incubated with primary antibody for 2 h at room temperature. Following three washes with 0.2% Tween 20 in PBS, pH 7.2 (PBST), the blots were incubated with horseradish peroxidase-conjugated secondary antibodies in blocking buffer for 1 h at room temperature. The blots were then washed three times with PBST, and developed with ECL (Amersham Pharmacia Biotech, Piscataway, NJ, USA) for 1 min. Band intensity was measured by Versa Doc 5000 (Bio Rad, Hercules, CA, USA).
Statistical analysis
Data were expressed as the mean ± SD. Differences in 4-OHE2 treated and non-treated group were analyzed by Student's t-test. Comparisons among the treatments were tested by ANOVA followed Duncan's multiple range test. All statistical tests were performed using the SAS statistical package Version 9.0 (SAS Institute, Cary, NC).
Results
Changes in oxidative stress markers
Following the exposure of cells to 0.1 µM 4-OHE2 for 96 h, intracellular ROS was increased by 60% (Fig. 1A). Cellular GSH/GSSG ratios were increased at 24 h and then decreased at 48 and 96 h (Fig. 1B). The expression of MnSOD, a key antioxidant enzyme involved in scavenging oxygen radicals was significantly decreased at 72 h and 96 h (Fig. 1C).
Changes in oxidative stress-related DNA damage response proteins
DNA damage response elements including BRCA1 and PARP-1 are closely related to the regulation of oxidative stress in breast cancer. As shown in Fig. 2A, BRCA1 protein levels were significantly suppressed at 48 h, while longer exposure (72 h) to 4-OHE2 restored the level. Fig. 2B shows a significant decrease in PARP-1 level at 48 h when cells were treated with 4-OHE2 treatment, while a considerable increase was observed at 72 h exposure.
Effects of tocopherols on modulating oxidative stress
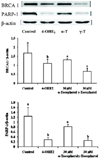
Cells treated with 30 µM γ-tocopherol showed a decreased level of intracellular ROS (Fig. 3A). γ-Tocopherol increased the GSH/GSSG ratio by reducing the level of GSSG (Fig. 3B). MnSOD protein level was increased with γ-tocopherol treatment (Fig. 3C). Although α-tocopherol did not exert a significant effect on these markers, it inhibited 4-OHE2-induced BRCA1 and PARP-1 depletion (Fig. 4A, 4B).
Discussion
Carcinogenesis can be viewed as a stepwise process beginning with genotoxic events followed by enhanced cell proliferation and tumor progression. Evidence has accumulated supporting the theory that catechol estrogens are oxidized to quinones accompanied by the generation of ROS, capable of causing genotoxicity and tumorigenicity. Despite the importance of this process, a limited number of studies have attempted to evaluate oxidized estrogen as a possible compound affecting redox balance in the cellular environment and the possible effects of antioxidant intervention. Therefore, we investigated the molecular events involved in 4-hydroxyestradiol (4-OHE2)-induced oxidative stresses as a possible events in cancer initiation process using normal breast epithelial cells. Protective roles of tocopherols were also determined.
In the present study, ROS levels, GSH/GSSG ratios, and manganese superoxide dismutase (MnSOD) protein levels were measured as indicators of oxidative stress in normal breast epithelial cells treated with 0.1 µM 4-OHE2, which is close to the physiological concentration in breast tissue. Changes in ROS level and the GSSG/GSH ratio indicate that intracellular ROS production caused by 4-OHE2 was possibly alleviated by GSH. In addition, reactive quinone formed from 4-OHE2 can produce a conjugate with GSH causing depletion of cellular thiol levels, which renders the cells vulnerable to oxidative damage. Since H2O2-detoxifying enzymes were poorly expressed in breast cancer cells (Esworthy et al., 1995), ROS accumulation may exert a greater effect in breast tumor development. We observed the restoration of GSH/GSSG balance in cells treated for 72 h. Cellular exposure to xenobiotics also results in a temporal increase in the total intracellular GSH content (Arrigo, 1999) indicating that cellular redox shift may stimulate GSH biosynthesis to overcome a redox shift, thereby stimulating GSH biosynthesis. Our study showed that MnSOD protein expression was decreased when cells were exposed to 4-OHE2 for more than 72 h, suggesting MnSOD protein expression is responsive to increased ROS levels caused by 4-OHE2 exposure. The decrease in MnSOD levels also leads to the accumulation of the superoxide anion (O2•) possibly inducing further oxidative damage.
BRCA1 and Poly (ADP-ribose) polymerase-1 (PARP-1) are established as DNA repair elements especially in breast tissue (Ginestier et al., 2009). Previous studies have reported that genetic polymorphisms in these genes are closely related to the increased risk of breast cancer (King et al., 2003; Lane, 1992; Malkin et al., 1990), indicating the functional importance of these genes in breast cancer development. It has been reported that estradiol and its metabolites induce mutation of chromosome 17 which contains the BRCA1 genes in human breast epithelial cells (Fernandez et al., 2006). Deficiency of BRCA1 and PARP-1 results in the accumulation of DNA strand breaks by impairing DNA damage repair, and causing defects in cell cycle checkpoints, centrosome amplification, and genetic instability (Brodie & Deng, 2001; Cantor & Andreassen, 2006; Deng & Wang, 2003; Venkitaraman, 2002; Zhang & Powell, 2005), which eventually result in tumorigenesis. Even though the effect was a transient, 4-OHE2 significantly decreased BRCA1 and PARP-1 protein expression. Since BRCA1 regulates the activity of nuclear factor (erythroid-derived 2)-like 2 (NFE2L2), an antioxidant response transcription factor, and is required for the repair of oxidative 8-oxoguanine lesions by transcription-coupled DNA repair (Bae et al., 2004), BRCA1 depletion by 4-OHE2 possibly accelerates carcinogenesis through inhibition of DNA damage repair and antioxidant enzyme expression.
Besides generating ROS during redox cycling, 4-OHE2 reacts with DNA to form covalently bound adducts such as 4-OHE2-N7-Gua and 4-OHE2-N3-Ade (Belous et al., 2007). ROS-induced oxidized bases are predominantly repaired via the base excision repair process (BER). It is generally believed that the repair of DNA single and double stranded breaks in mammalian cells is initiated by PAPR-1 (de Murcia & Ménissier de Murcia, 1994; Plummer, 2006). Moreover, PARP-1 has a functional domain that includes the BRCA1 C-terminal domain (Plummer, 2006), and therefore PARP-1 deficiency impairs BRCA1 recruitment to the site of DNA damage (De Soto & Deng, 2006), which may facilitate cancer development. A reduction in PARP-1 protein expression and an increase in its activity at 48 and 96 h were possibly accompanied by 4-OHE2-induced ROS production. Chen et al. (2005) also showed that treatment with 4-OHE2 at 25 µM for 24 h causes PARP-1 cleavage. Depletion of both PARP-1 and BRCA1 at 48 h can therefore cause an overload of 4-OHE2 induced DNA damage.
Since exogenous antioxidants may regulate 4-OHE2-induced cellular oxidative stress, we next evaluated the effects of tocopherols on oxidative stress markers and DNA repair protein expression. In these experiments, we used tocopherols at 30 µM which falls within the normal range of blood level in human. For measurement of oxidative stress markers, 4-OHE2 and/or tocopherols were treated for 96 h, because our pretests indicated that the disturbance of antioxidatve system was induced within 96 h. Study results indicated that in 4-OHE2 treated MCF-10A cells, the accumulation of intracellular ROS and depletion of GSH and MnSOD were significantly attenuated by γ-tocopherol. The antioxidant activity of tocopherols is rooted in their ability to donate phenolic hydrogens to lipid radicals. Because it lacks electron-donating methyl groups on the chromanol ring, γ-tocopherol is somewhat less potent in donating electrons than α-tocopherol and thus a slightly less powerful antioxidant. However, the unsubstituted C-5 position of γ-tocopherol appears to make it as a better trap for lipophilic electrophiles such as reactive nitrogen oxide species (RNOSs). Jiang et al. (2002) showed that in vivo γ-tocopherol supplementation, not α-tocopherol, resulted in inhibition of protein nitration and ascorbate oxidation in kidney. Li et al. (1999) also reported that γ-tocopherol was more potent than α-tocopherol in enhancing MnSOD protein expression in arterial tissues affected by cardiovascular disease. Mense et al. (2009) showed that antioxidant vitamin C reduces the incidence of estrogen-induced mammary tumors and increases tumor latency through changes in the activities of antioxidant enzymes superoxide dismutase and glutathione peroxidase. The cellular functions of α-tocopherol that are independent of this antioxidant/radical scavenging ability have been well characterized. A recent report suggests that vitamin E isomers possess anti-cancer activity by regulating DNA repair gene and apoptosis via signaling pathways (Aiub et al., 2009; Theriaut et al., 1999). Therefore, we determined the effects of tocopherols on DNA repair elements in 4-OHE2 induced oxidative stress in breast epithelial cells. We treated 4-OHE2 and/or tocopherols for 48 h because our pretests indicated that 4-OHE2 induced the depletion of BRCA1 and PARP-1 protein expression at 48 hr. Our results showed that α-tocopherol stimulates the up-regulated expression of DNA repair proteins, including BRCA1 and PARP-1. Yu et al. (2003) and Gu et al. (2008) reported that human cancer cells undergo apoptosis stimulated by α-tocopherol succinate in a concentration- and time-dependent manner by restoring PARP-1 levels.
In conclusion, the estrogen metabolite, 4-OHE2 affects cellular redox status, regulates the expression of proteins closely related DNA repair and promotes breast cancer cell growth. As far as we know, this is one of few studies to examine the involvement of oxidized estradiol in the impaired DNA repair. Exogenous α- and γ-tocopherol played a role as anti-cancer compounds by modulating 4-OHE2 induced oxidative stresses.
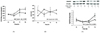


 XML Download
XML Download