

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Angelica keiskei (AK) is a large perennial herb belongings to the Umbelliferae family. It grows mainly along the Pacific coast of Asia and is known in Japan as ashitaba and in Korea as sinsuncho [1]. AK is widely cultivated as a functional food and beverage ingredients. AK green tea is widely consumed in Asian countries including China, India, and Japan as a health-related drink. AK leaves have been consumed as food and medicine in Japan and Korea [2]. Several studies have shown that AK possesses a wide range of pharmacologic and health-promoting properties including anti-inflammatory [23], anti-adiposity [4], anti-diabetic, anti-hypertensive [5], and anti-tumor effects [67]. Many of these properties have been described as resulting from various active chalcones, coumarines and flavonoids in AK [126]. In addition, AK has widely been utilized among Koreans that believe it can protect against hepatotoxicity. Noh et al. [8] recently reported that an AK extract supplement can improve liver function in heavy drinkers. However, the effects of AK on hepatotoxicity and its mechanisms of action have yet to be elucidated.
Worldwide, acetaminophen (AAP) is widely prescribed for treatment of mild pain and fever. AAP is generally considered a safe and effective anesthetic and antipyretic drug at therapeutic doses; however, an AAP overdose can result in severe liver injury through mitochondrial dysfunction and damage [910]. AAP overdose is the most common cause of acute liver failure in most western countries [11]. Several studies have revealed that AAP causes apoptosis in hepatocytes and then exhibits hepatotoxicity under in vitro and in vivo conditions [11]. N-acetyl-p-benzoquinone imine, the toxic metabolite of AAP, inhibits mitochondrial oxidative phosphorylation, depletes adenosine triphosphate (ATP), produces selective mitochondrial oxidant stress [12], decreases mitochondrial membrane potential, increases Ca2+ levels, changes membrane permeability transition, and releases pro-apoptotic factors into the cytosol [13]. These actions are accompanied by caspase-3 activation, DNA fragmentation, and apoptotic cell death [14]. AAP-induced hepatotoxicity has been used as a useful model in hepatoprotective study.
In this study, we attempted to investigate the protective effects of an AK ethanol extract (AK-Ex) against AAP-induced liver injury in HepG2 human hepatocellular liver carcinoma cells and HepaRG human hepatic progenitor cells. We also examined the possible molecular mechanisms of their activities.
MATERIALS AND METHODS
Materials
Anti-β-actin antibody and 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco's modified Eagle's medium (DMEM) was purchased from Gibco BRL (Gaithersburg, MD, USA). Fetal bovine serum (FBS), trypsin/EDTA and penicillin/streptomycin were purchased from Cambrex Bio Technology (Walkersville, MD, USA). Antibodies against cleaved caspase-3, cleaved caspase-7, cleaved caspase-9, and cleaved poly (ADP-ribose) polymerase (PARP) were purchased from Cell Signal Technology (Beverly, MA, USA). Antibodies against Bcl-2, Bcl-xL, Bax, Bak, Bok, and Bik were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Preparation of extract
Dried AK cultivated in Chuncheon, Korea was purchased from an herbal medicine store in Seoul, Korea. Dried AK was pulverized by using a blender. The powder (1 kg) was refluxed in 1 L of 70% ethanol at 100℃ for 8 h. The supernatant solution was filtered through Whatman filter paper #2, after which the filtrate was evaporated in a rotary vacuum evaporator and subsequently freeze-dried at −70℃. The resulting powder was used as an ethanol extract of AK (AK-Ex) and preserved at −20℃ for use. The yield of AK-Ex was 241.6 g per kilogram of dried AK powder.
Cell culture
The HepG2 human hepatoma cell line was obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in DMEM supplemented with 100 mL/L FBS, 100,000 U/L penicillin, and 100 mg/L streptomycin. HepaRG human hepatic progenitor cells were obtained from Life Technology (Carlsbad, CA, USA). HepaRG cells were cultured in Williams' medium E (WME) supplemented with 200 mL/L HepaRG™ Tox medium supplement and 10 mL/L GlutaMAX™ supplement according to the recommendations of the manufacturer (Life Technology). HepG2 and HepaRG cells were maintained in an incubator at 37℃ in a humidified atmosphere of 5% CO2 and 95% air.
Cell viability assay
In order to assess the effect of AK-Ex on the growth of HepG2 and HepaRG cells, cells were plated in multi-well plates with their respective culture media. After incubation for 24 h, we replaced the respective culture media with various concentrations of AK-Ex and/or 30 mM AAP as indicated. Viable cell numbers were estimated via MTT assays, as previously described [15].
Lactate dehydrogenase assay
Leakage of lactate dehydrogenase (LDH) was measured as an index of cell membrane damage [16]. HepG2 cells were plated at a density of 5 × 104 cells/well in a 24-well plate and incubated for 24 h. HepaRG cells were plated at a density of 1.2 × 105 cells/well in a 24-well plate and incubated for 72 h. Cells were treated for 48 h with various concentration of AK-Ex and then treated with 30 mM AAP for an additional 24 h. The 24 h-conditioned media were collected for use in LDH activity assays. The LDH activity was measured by using an LDH cytotoxicity detection assay kit (Takara Bio Inc., Osaka, Japan) according to the manufacturer's instructions.
Quantification of apoptotic cells and measurement of mitochondrial membrane depolarization
HepG2 cells and HepaRG cells were treated with AK-Ex and/or AAP, respectively, as described above. Apoptotic cells were quantified by using a cell death detection ELISAPLUS assay kit (Roche Applied Science, Switzerland) in accordance with the manufacturers' instructions. To measure mitochondrial membrane depolarization, a dual-emission, potential-sensitive probe JC-1 was utilized as described previously [17].
Western blot analysis
Cells were lysed as described previously [18]. The protein contents of the cell lysates were measured by using a BCA protein assay kit (Pierce, Rockford, IL, USA). Western blot analyses were conducted as described previously [18]. Blots were developed by using Immobilon™ Western chemiluminescent HRPsubstrate (Pierce). The intensity of each band was quantified by using the Bio-profile Bio-1D application (Vilber, Eberhadzell, Germany). The expressions were normalized to β-actin.
Statistical analysis
Results are expressed as means ± SEM values. Significant differences between means were determined by performing analysis of variance. Differences between groups were determined by using Duncan's multiple range test. Analyses utilized the SAS statistical analysis software package (SAS Institute, Cary, NC, USA). Differences were regarded significant at P < 0.05.
RESULTS
Effect of an ethanol extract of Angelica keiskei (AK-Ex) on viable cell numbers against acetaminophen (AAP)-induced hepatotoxicity in HepG2 and HepaRG cells
We first assessed the effect of various concentrations (0, 50, 75, 100 µg/mL) of AK-Ex on cell growth against AAP-induced hepatotoxicity in HepG2 cells by estimating the total number of viable cells by performing MTT assays. As shown in Fig. 1, according to the concentration of AK-Ex and under AAP overdose treatment, the effects on cell growth against AAP-induced hepatotoxicity were different. When treating AK-Ex and AAP simultaneously or when inducing hepatotoxicity first by treating with of AAP overdose and subsequently treating AK-Ex, there were significant differences in the number of viable cells. However, there was a remarkably marked increase in the number of viable cells when treating AK-Ex first and then inducing AAP-hepatotoxicity (Fig. 1). Therefore, AK-Ex seems to be effective in preventing AAP-induced hepatotoxicity when HepG2 cell are pretreated with AK-Ex. To examine the effect on protecting liver cell against AAP-induced hepatotoxicity according to AK-Ex pretreatment time, HepG2 cells in culture media containing various concentrations of AK-Ex were treated for 24, 36, and 48 h followed by 30 mM AAP to induce hepatotoxicity. The number of viable cells were determined after 24 h. The results show that AK-Ex increased the number of viable cell numbers in a time-dependent manner (Fig. 2A).
The HepG2 cell line originated from a human hepatocellular carcinoma and has been a useful human cellular liver model for toxicity testing. One of the reasons for its usefulness is that it is easy to cultivate HepG2 cells [19]. Although the HepG2 cell line indicates many liver-specific functions, it also has some disadvantages, the most distinct being that in comparison to primary human hepatocytes, HepG2 cells differ in the expression and activity levels of many xenobiotic metabolizing enzymes and transporters [19202122]. Therefore, we examined the effects of AK-Ex on cell growth in HepaRG cells, the primary human hepatocyte. It is hard to use HepaRG for study because of its need for difficult culture conditions. However, HepaRG may represent a more promising liver model than HepG2 when evaluating hepatic drug metabolism under in vitro conditions because HepaRG possesses the metabolic capacity characteristics of primary human hepatocytes [232425].
As shown in Fig. 2B, after 24, 36, 48 h of Ak-Ex pretreatment in HepaRG cells, we induced hepatotoxicity by using 30 mM AAP and measured the number of viable cells after 24 h. As a result, the number of viable cell significantly increased in HepaRG cells, as was also observed in HepG2 cells, as pretreatment time increased. Therefore, AK-Ex can reduce AAP-induced hepatotoxicity in HepaRG cells, as well as in HepG2 cells.
Effect of AK-Ex on lactate dehydrogenase release in HepG2 and HepaRG cells intoxicated with AAP
LDH was found in all cells and its release was regarded as a biomarker of cell cytotoxicity, and such relases increases mitochondrial permeability and damage [26]. In this study, in order to determine whether AK-Ex changes the LDH concentration in culture media, we measured the LDH concentration of HepG2 and HepaRG cells by culturing with various concentrations of AK-Ex for 48 h and then culturing for another 24 h after adding 30 mM AAP. As shown in Fig. 3A and 3B, AK-Ex reduced the membrane permeability resulting in AAP-induced hepatotoxicity in HepG2 and HepaRG cells in a dose-dependent manner. These results show that pretreatment of HepG2 and HepaRG cells with AK-Ex can prevent AAP-induced hepatotoxicity.
Effect of AK-Ex on apoptosis in HepG2 and HepaRG cells intoxicated with AAP
To investigate the effect of AK-Ex on apoptosis in hepatocytes, we examined the effect of AK-Ex on DNA fragmentation. HepG2 or HepaRG cells were treated with 0, 50, 75, 100 µg/mL AK-Ex and then cytotoxicity was induced by 30 mM AAP treatment. DNA fragmentation, a morphometric characteristics of apoptosis was detected via a cell death detection ELISAPLUS assay kit. As shown in Fig. 4A and 4B, the number of apoptotic cell increased following AAP overdose in HepG2 and HepaRG cell lines. However, when the cells were pretreated with AK-Ex for 48 h, AAP-induced apoptosis significantly decreased in a dose-dependent manner. Thus, AK-Ex pretreatment can reduce AAP-induced hepatotoxicity.
Effect of AK-Ex on depolarization of mitochondria in HepG2 cells intoxicated with AAP
To determine whether AK-Ex-reduced apoptosis against AAP-induced cytotoxicity was mediated via a mitochondrial-dependent pathway, we used JC-1 as a mitochondrial depolarization marker. JC-1 was used because of its capability to detect changes in the mitochondrial membrane potential of living cells. As shown in Fig. 5, AK-Ex increased the percentage of cells with intact mitochondria (red positive and green negative) and decreased the percentage of cells with depolarized mitochondrial membranes (green positive and red negative) in an AK-Ex concentration-dependent manner. The results show that mitochondrial membrane permeability decreases in HepG2 cells against AAP-induced hepatotoxicity by pretreatment of AK-Ex in a dose-dependent manner.
Effect of AK-Ex on the levels of Bcl-2 family proteins in HepG2 cells intoxicated with AAP
To explain the molecular mechanism of AK-Ex associated with the suppression of apoptosis, we examined the effects of AK-Ex on the expression of Bcl-2 family proteins in AAP-induced hepatotoxic HepG2 cells. Bcl-2 family proteins regulate apoptosis by controlling mitochondrial membrane permeability and cytochrome c release [27]. Cells were treated with various concentrations of AK-Ex for 48 h and then treated with 30 mM AAP, after which total cell lysates were prepared for western blot analysis. We analyzed the protein expressions of Bax, Bok, Bik, and Bak as proapoptotic markers and Bcl-2 and Bcl-xL as anti-apoptotic markers. AK-Ex increased the levels of the anti-apoptotic Bcl-2 protein but did not affect Bcl-xL expression in AAP-induced hepatotoxic HepG2 cells. AK-Ex did not affect the levels of the proapoptotic protein Bak, but Bax, Bok, and Bik expressions were decreased in cells treated with AK-Ex (Fig. 6).
Effect of AK-Ex on activation of caspases and PARP cleavage in HepG2 and HepaRG cells intoxicated with AAP
We also determined whether AK-Ex activates caspases. To that end, we performed western blotting with antibodies that detect cleaved forms of the enzymes. AK-Ex decreased the cleaved caspase-9, -7 and -3 levels in concentration-dependent manners. In addition, the PARP cleavage product significantly decreased with increasing concentrations of AK-Ex (Fig. 7A). We then tried to determine whether AK-Ex affects the PARP cleavage in HepaRG cells. The results showed that the PARP cleavage product significantly decreased with increasing concentrations of AK-Ex (Fig. 7B).
DISCUSSION
AK has been used as a folk medicine and as a health-promoting herb. It has been reported that AK contains numerous bioactive substances such as chalcones, coumarines, flavonoids, saponins, and vitamins and exhibits anti-inflammatory [2328], anti-adiposity [429], anti-oxidant [30], anti-diabetic [3132], anti-hypertensive [5], anti-tumor [67], and anti-memory impairment [33] effects.
In the present study, we found that AK-Ex treatment, when executed prior to AAP treatment, increased the total viable cell numbers in HepG2 cells against AAP-induced hepatotoxicity (Fig. 1). Therefore, AK-Ex appears to effectively protect against future hepatotoxicity if, and only if, it is used as a supplement in advance of hepatic damage. Other researchers have also reported the hepatoprotective effect of AK [34]. An AK methanol extract, when administered prior to galactosamine, exerted protective effects against galactosamine-induced hepatotoxicity in rat [34]. These results suggest that AK has a hepatoprotective effects, but such effects are not to be equally expected as they depend on the hepatotoxines employed. In addition, our results indicate that AK-Ex reduces apoptosis against AAP-induced hepatotoxicity in HepG2 and HepaRG cells.
Apoptosis is an important process resulting in programmed cell death. Activation of apoptosis is initiated via two main pathways: intrinsic and/or extrinsic [35]. Mitochondria are an essential mediator of intrinsic apoptosis, and the Bcl-2 family of proteins, located in the outer mitochondrial membrane and consisting of proapoptotic (such as Bax, Bid, Bik, Bak, Bok) and antiapoptotic mediators (such as Bcl-2, Bcl-xL, Mcl-1), is important in modulating apoptosis by its ability to alter mitochondrial permeability to apoptotic mediators [36]. The permeability of the mitochondrial membrane is crucial in several apoptosis pathways. Apoptosis in mammalian cells has been shown to be regulated by Bcl-2 family of proteins [37]; thus, we determined whether AK-Ex has a protective effect on AAP-induced apoptosis in HepG2 cells and examined its association with the modulation of these proteins. AK-Ex, in a concentration-dependent manner, significantly reduced apoptosis and the percentage of cells with depolarized mitochondrial membranes, and it increased the percentage of cells with intact mitochondria (Fig. 4, 5). Western blotting results indicated that AK-Ex treatment increased the expression of Bcl-2 and decreased the expression of Bax, Bok, and Bik; however, it did not change the expressions of antiapoptotic Bcl-xL or pro-apoptotic Bak (Fig. 6). Bax and Bcl-2 are important regulators of cytochrome c release from mitochondria [37]. Internal damage to a cell causes a decrease in the Bcl-2 / Bax ratio, which results in holes in the mitochondrial membrane. These inter-mitochondrial apoptotic molecules are released into the cytosol upon various forms of signals, leading to changes in permeability of the mitochondrial membrane [38]. Cytochrome c is one of the pro-apoptotic molecules that, leads to activation of caspases and disrupts outer mitochondrial membrane permeability. In the present study, AK-Ex significantly increased the Bcl-2 / Bax ratio against AAP-induced cytotoxicity in HepG2 cells. Thus it seems that AK-Ex-inhibited apoptosis involves Bax and Bcl-2 signal transduction.
Activation of apoptosis via the extrinsic pathway occurs via the binding of apoptotic ligands to death receptors [39]. These death receptors have an intracellular domain that functions as a protein-binding module. Following the recruitment and signaling of various adaptor molecules, cleavage and activation of initiator caspase-8 -9, -10, and -12 occurs. In turn, this leads to activation of executioner caspase-3, -6, and -7 as well as effector caspases, culminating in DNA fragmentation [40]. In the present study, AK-Ex decreased cleaved caspase-9, -7, and -3 and cleaved PARP (Fig. 7). AK-Ex activates caspase-9, a cysteine protease, and caspase-9 activation lead to activation of caspase-7 and caspase-3, which are responsible for destroying the cell from within.
PARP found in the cell's nucleus, is a family of proteins involved in DNA repair and programmed cell death. PARP can be activated in cells experiencing stress and/or DNA damage, and PARP is inactivated by caspase-3 cleavage during programmed cell death. Cleaved PARP is a marker for apoptosis [41]. In this study, AK-Ex treatment decreased cleaved PARP in HepG2 and HepaRG cells intoxicated by AAP (Fig. 7). Therefore, the present study's results suggest that AK-Ex suppresses AAP-induced hepatocytotoxic apoptosis via both intrinsic and extrinsic pathways in HepG2 and HepaRG cells.
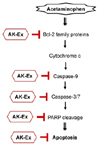
In the present study, we show that AK-Ex suppresses apoptosis; decreases mitochondrial membrane permeability; increases anti-apoptotic mediators such as Bcl-2, decreases pro-apoptotic mediators such as Bax, Bok, and Bik, and decreases cleaved caspase-9, -7, and -3, and cleaved PARP. Taken together, the present results suggest that AK-Ex suppresses AAP-induced hepatocytotoxic apoptosis via both intrinsic and extrinsic pathway in HepG2 and HepaRG cells (Fig. 8).
In conclusion, our data demonstrated that AK-Ex protects againsted AAP-induced growth inhibition and apoptosis induction in HepG2 and HepaRG cells. Moreover, AK-Ex modulates the expression of Bcl-2 family proteins and decreases the levels of cleaved caspase-9, -7, and -3, and cleaved PARP under AAP-induced hepatotoxicity. Based on these observations, we suggest that AK could be a useful hepatoprotective agent against AAP-induced apoptosis in hepatocytes. Future in vivo studies of animal hepatotoxic models are needed to examine whether AK-Ex could be a useful hepatoprotective treatment agent against hepatotoxicity.







 XML Download
XML Download