1. Lim SS, Vos T, Flaxman AD, Danaei G, Shibuya K, Adair-Rohani H, Amann M, Anderson HR, Andrews KG, Aryee M, Atkinson C, Bacchus LJ, Bahalim AN, Balakrishnan K, Balmes J, Barker-Collo S, Baxter A, Bell ML, Blore JD, Blyth F, Bonner C, Borges G, Bourne R, Boussinesq M, Brauer M, Brooks P, Bruce NG, Brunekreef B, Bryan-Hancock C, Bucello C, Buchbinder R, Bull F, Burnett RT, Byers TE, Calabria B, Carapetis J, Carnahan E, Chafe Z, Charlson F, Chen H, Chen JS, Cheng AT, Child JC, Cohen A, Colson KE, Cowie BC, Darby S, Darling S, Davis A, Degenhardt L, Dentener F, Des Jarlais DC, Devries K, Dherani M, Ding EL, Dorsey ER, Driscoll T, Edmond K, Ali SE, Engell RE, Erwin PJ, Fahimi S, Falder G, Farzadfar F, Ferrari A, Finucane MM, Flaxman S, Fowkes FG, Freedman G, Freeman MK, Gakidou E, Ghosh S, Giovannucci E, Gmel G, Graham K, Grainger R, Grant B, Gunnell D, Gutierrez HR, Hall W, Hoek HW, Hogan A, Hosgood HD 3rd, Hoy D, Hu H, Hubbell BJ, Hutchings SJ, Ibeanusi SE, Jacklyn GL, Jasrasaria R, Jonas JB, Kan H, Kanis JA, Kassebaum N, Kawakami N, Khang YH, Khatibzadeh S, Khoo JP, Kok C, Laden F, Lalloo R, Lan Q, Lathlean T, Leasher JL, Leigh J, Li Y, Lin JK, Lipshultz SE, London S, Lozano R, Lu Y, Mak J, Malekzadeh R, Mallinger L, Marcenes W, March L, Marks R, Martin R, McGale P, McGrath J, Mehta S, Mensah GA, Merriman TR, Micha R, Michaud C, Mishra V, Mohd Hanafiah K, Mokdad AA, Morawska L, Mozaffarian D, Murphy T, Naghavi M, Neal B, Nelson PK, Nolla JM, Norman R, Olives C, Omer SB, Orchard J, Osborne R, Ostro B, Page A, Pandey KD, Parry CD, Passmore E, Patra J, Pearce N, Pelizzari PM, Petzold M, Phillips MR, Pope D, Pope CA 3rd, Powles J, Rao M, Razavi H, Rehfuess EA, Rehm JT, Ritz B, Rivara FP, Roberts T, Robinson C, Rodriguez-Portales JA, Romieu I, Room R, Rosenfeld LC, Roy A, Rushton L, Salomon JA, Sampson U, Sanchez-Riera L, Sanman E, Sapkota A, Seedat S, Shi P, Shield K, Shivakoti R, Singh GM, Sleet DA, Smith E, Smith KR, Stapelberg NJ, Steenland K, Stöckl H, Stovner LJ, Straif K, Straney L, Thurston GD, Tran JH, Van Dingenen R, van Donkelaar A, Veerman JL, Vijayakumar L, Weintraub R, Weissman MM, White RA, Whiteford H, Wiersma ST, Wilkinson JD, Williams HC, Williams W, Wilson N, Woolf AD, Yip P, Zielinski JM, Lopez AD, Murray CJ, Ezzati M, AlMazroa MA, Memish ZA. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990-2010: a systematic analysis for the Global Burden of Disease Study 2010. Lancet. 2012; 380:2224–2260. PMID:
23245609.

2. Eckel RH, Grundy SM, Zimmet PZ. The metabolic syndrome. Lancet. 2005; 365:1415–1428. PMID:
15836891.
3. Seppälä-Lindroos A, Vehkavaara S, Häkkinen AM, Goto T, Westerbacka J, Sovijärvi A, Halavaara J, Yki-Järvinen H. Fat accumulation in the liver is associated with defects in insulin suppression of glucose production and serum free fatty acids independent of obesity in normal men. J Clin Endocrinol Metab. 2002; 87:3023–3028. PMID:
12107194.
4. Kotronen A, Westerbacka J, Bergholm R, Pietiläinen KH, Yki-Järvinen H. Liver fat in the metabolic syndrome. J Clin Endocrinol Metab. 2007; 92:3490–3497. PMID:
17595248.
5. Ridker PM, Buring JE, Cook NR, Rifai N. C-reactive protein, the metabolic syndrome, and risk of incident cardiovascular events: an 8-year follow-up of 14 719 initially healthy American women. Circulation. 2003; 107:391–397. PMID:
12551861.
6. Mertens I, Verrijken A, Michiels JJ, Van der Planken M, Ruige JB, Van Gaal LF. Among inflammation and coagulation markers, PAI-1 is a true component of the metabolic syndrome. Int J Obes (Lond). 2006; 30:1308–1314. PMID:
16389265.
7. Park EJ, Lee JH, Yu GY, He G, Ali SR, Holzer RG, Österreicher CH, Takahashi H, Karin M. Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010; 140:197–208. PMID:
20141834.
8. Choi SW, Friso S. Epigenetics: a new bridge between nutrition and health. Adv Nutr. 2010; 1:8–16. PMID:
22043447.
9. Milagro FI, Campión J, García-Díaz DF, Goyenechea E, Paternain L, Martínez JA. High fat diet-induced obesity modifies the methylation pattern of leptin promoter in rats. J Physiol Biochem. 2009; 65:1–9. PMID:
19588726.
10. Jiang M, Zhang Y, Liu M, Lan MS, Fei J, Fan W, Gao X, Lu D. Hypermethylation of hepatic glucokinase and L-type pyruvate kinase promoters in high-fat diet-induced obese rats. Endocrinology. 2011; 152:1284–1289. PMID:
21239437.
11. Schwenk RW, Jonas W, Ernst SB, Kammel A, Jähnert M, Schürmann A. Diet-dependent alterations of hepatic Scd1 expression are accompanied by differences in promoter methylation. Horm Metab Res. 2013; 45:786–794. PMID:
23803969.
12. Chang X, Yan H, Fei J, Jiang M, Zhu H, Lu D, Gao X. Berberine reduces methylation of the MTTP promoter and alleviates fatty liver induced by a high-fat diet in rats. J Lipid Res. 2010; 51:2504–2515. PMID:
20567026.
13. Dudley KJ, Sloboda DM, Connor KL, Beltrand J, Vickers MH. Offspring of mothers fed a high fat diet display hepatic cell cycle inhibition and associated changes in gene expression and DNA methylation. PLoS One. 2011; 6:e21662. PMID:
21779332.
14. Zhang Y, Wang H, Zhou D, Moody L, Lezmi S, Chen H, Pan YX. High-fat diet caused widespread epigenomic differences on hepatic methylome in rat. Physiol Genomics. 2015; 47:514–523. PMID:
26199400.
15. Richardson B. Impact of aging on DNA methylation. Ageing Res Rev. 2003; 2:245–261. PMID:
12726774.
16. Folch J, Lees M, Sloane Stanley GH. A simple method for the isolation and purification of total lipides from animal tissues. J Biol Chem. 1957; 226:497–509. PMID:
13428781.
17. Jin SG, Kadam S, Pfeifer GP. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res. 2010; 38:e125. PMID:
20371518.
18. Irizarry RA, Ladd-Acosta C, Carvalho B, Wu H, Brandenburg SA, Jeddeloh JA, Wen B, Feinberg AP. Comprehensive high-throughput arrays for relative methylation (CHARM). Genome Res. 2008; 18:780–790. PMID:
18316654.
19. Tammen SA, Park LK, Dolnikowski GG, Ausman LM, Friso S, Choi SW. Hepatic DNA hydroxymethylation is site-specifically altered by chronic alcohol consumption and aging. Eur J Nutr. Forthcoming 2015.
20. McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010; 28:495–501. PMID:
20436461.
21. Fujita PA, Rhead B, Zweig AS, Hinrichs AS, Karolchik D, Cline MS, Goldman M, Barber GP, Clawson H, Coelho A, Diekhans M, Dreszer TR, Giardine BM, Harte RA, Hillman-Jackson J, Hsu F, Kirkup V, Kuhn RM, Learned K, Li CH, Meyer LR, Pohl A, Raney BJ, Rosenbloom KR, Smith KE, Haussler D, Kent WJ. The UCSC Genome Browser database: update 2011. Nucleic Acids Res. 2011; 39:D876–D882. PMID:
20959295.
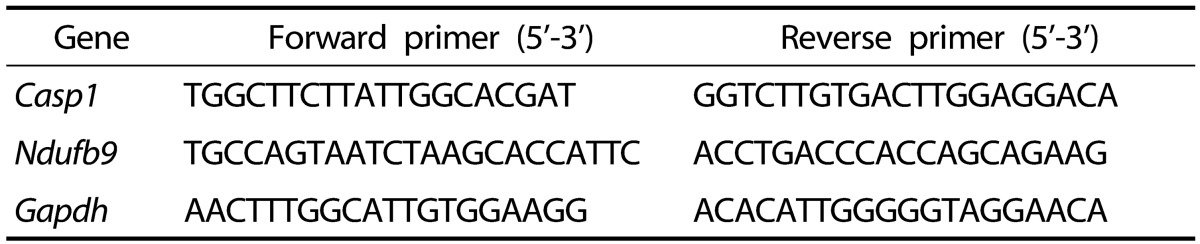
22. Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) method. Methods. 2001; 25:402–408. PMID:
11846609.
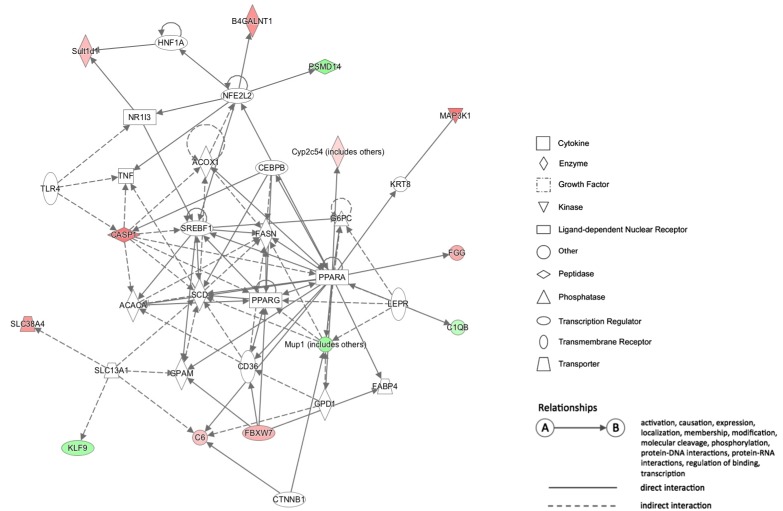
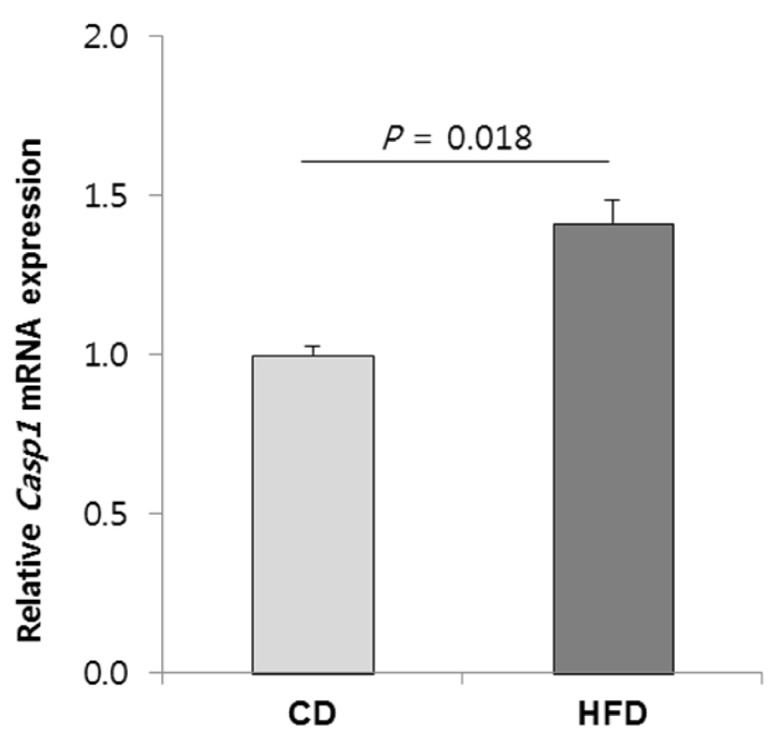
23. Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 2009; 10:241–247. PMID:
19221555.
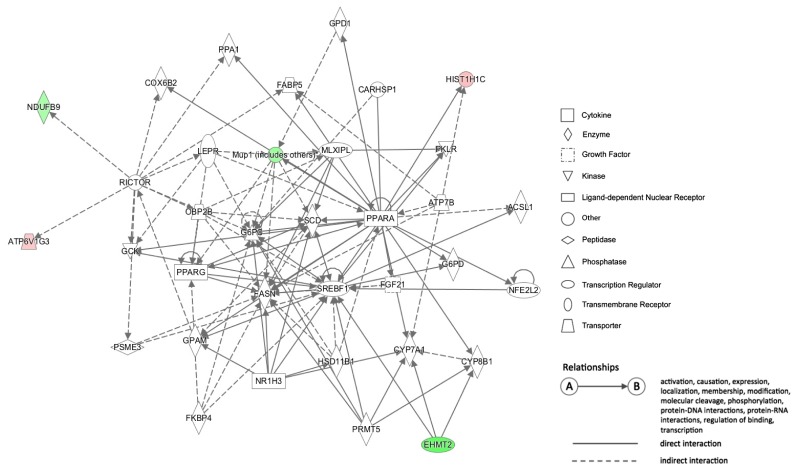
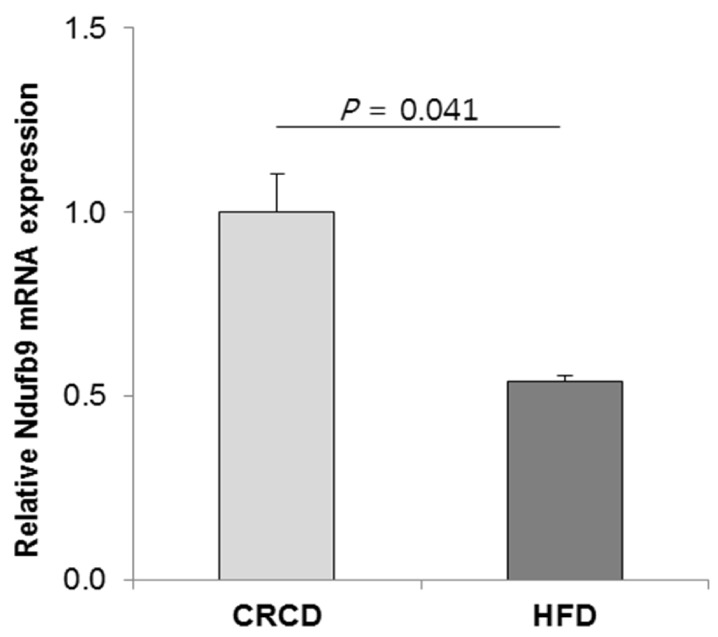
24. Smeitink J, van den Heuvel L. Human mitochondrial complex I in health and disease. Am J Hum Genet. 1999; 64:1505–1510. PMID:
10330338.
25. Haack TB, Madignier F, Herzer M, Lamantea E, Danhauser K, Invernizzi F, Koch J, Freitag M, Drost R, Hillier I, Haberberger B, Mayr JA, Ahting U, Tiranti V, Rötig A, Iuso A, Horvath R, Tesarova M, Baric I, Uziel G, Rolinski B, Sperl W, Meitinger T, Zeviani M, Freisinger P, Prokisch H. Mutation screening of 75 candidate genes in 152 complex I deficiency cases identifies pathogenic variants in 16 genes including NDUFB9. J Med Genet. 2012; 49:83–89. PMID:
22200994.
26. Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012; 13:484–492. PMID:
22641018.
27. Ge ZJ, Luo SM, Lin F, Liang QX, Huang L, Wei YC, Hou Y, Han ZM, Schatten H, Sun QY. DNA methylation in oocytes and liver of female mice and their offspring: effects of high-fat-diet-induced obesity. Environ Health Perspect. 2014; 122:159–164. PMID:
24316659.
28. Murphy SK, Yang H, Moylan CA, Pang H, Dellinger A, Abdelmalek MF, Garrett ME, Ashley-Koch A, Suzuki A, Tillmann HL, Hauser MA, Diehl AM. Relationship between methylome and transcriptome in patients with nonalcoholic fatty liver disease. Gastroenterology. 2013; 145:1076–1087. PMID:
23916847.
29. Choi MS, Kim YJ, Kwon EY, Ryoo JY, Kim SR, Jung UJ. High-fat diet decreases energy expenditure and expression of genes controlling lipid metabolism, mitochondrial function and skeletal system development in the adipose tissue, along with increased expression of extracellular matrix remodelling- and inflammation-related genes. Br J Nutr. 2015; 113:867–877. PMID:
25744306.
30. Jones PA. The DNA methylation paradox. Trends Genet. 1999; 15:34–37. PMID:
10087932.
31. Lorincz MC, Dickerson DR, Schmitt M, Groudine M. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol. 2004; 11:1068–1075. PMID:
15467727.
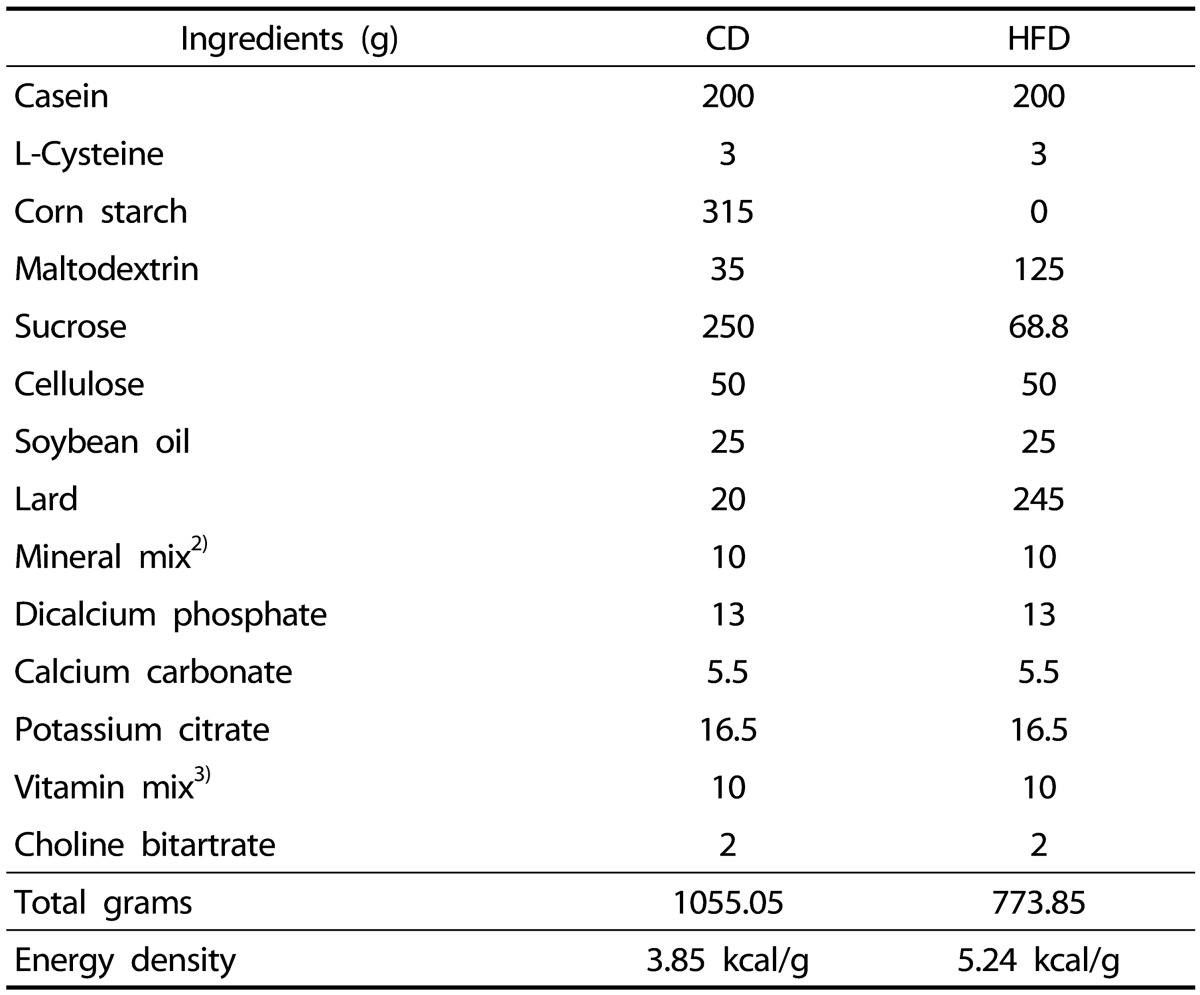
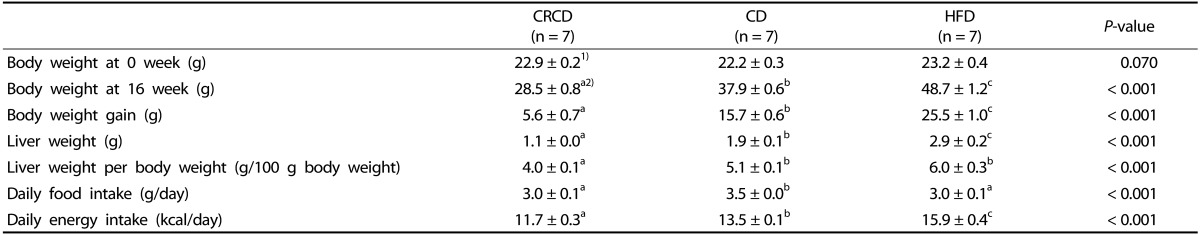
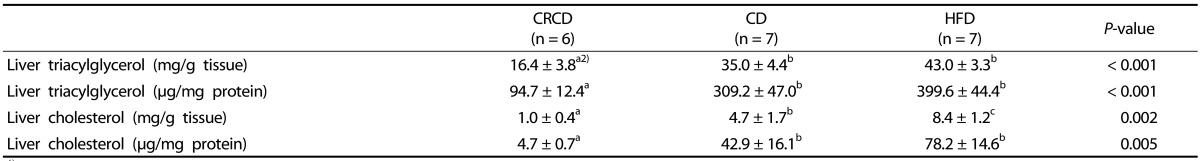
32. National Research Council (US) Subcommittee on Laboratory Animal Nutrition. Nutrient Requirements of Laboratory Animals. 4th rev. ed. Washington (D.C.): National Academies Press;1995.


 PDF
PDF ePub
ePub Citation
Citation Print
Print


 XML Download
XML Download