

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Epstein Barr virus (EBV) is herpes virus type 4 which was first reported by Epstein and Barr at 1964 and over 95% of all adults were infected with EBV. This virus infects not only specifically human B cells through surface CD21 or MHC molecules, but also T cell, follicular dendritic cell and epithelial cells in a low probability. When EBV infects young children, it causes a minor cold-like illness or asymptomatic responses. However, in adolescents and adults, EBV may cause infectious mononucleosis (1,2). In addition, EBV is deeply associated with several tumors including Burkitt's lymphoma Hodgkin's disease, nasopharyngeal carcinoma, and gastric carcinoma (3,4). Because EBV is so common and widespread, preventing its infection is almost impossible. Currently, there is no specific treatment to EBV. Thus it is important to regulate or modulate EBV-infected cells in living person, many scientists have been studying about that. When EBV enters human B cells, it conducts either lytic program or latency program. Latency program is established in most people having EBV infection. EBV infects human B cells in vitro, it can transform B cells into lymphoblastoid cell lines which are permanently growing. The transformed B cells are good models for B cell activation, differentiation, and B cell tumorogenesis (5,6). EBV-transformed B cells express several activation markers such as CD38, CD77 and costimulatory molecules such as CD80 and CD86.
B7 family is representative costimulatory molecule found on activated antigen presenting cells. There are seven known members of the B7 family, B7.1 (CD80), B7.2 (CD86), programmed death (PD)-L1, PD-L2, B7-H3, B7-H4, and inducible costimulatory ligand (ICOS-L). CD80 and CD86 are the most well-known B7 molecules. They have different functions depending on receptors of coupling. When CD80 or CD86 interacts with surface CD28 on T cells together with antigen-TCR (T cell receptor) binding, they can activate resting T cell. However, they also inhibit activation of T cell, interacting with CTLA-4 (CD152) on activated T cell (7-10). Thus most studies about functions of CD80 and CD86 have focused on the activation and regulation of T cells through CD28 or CTLA-4, but downstream signal of CD80 and CD86 is not fully understood. As B cell is actually one of antigen presenting cells, resting B cell have weak expression of CD86 and no expression of CD80 (11-14). B cell receptor signal induced abundant expression of CD86. Lipopolysaccharide, CD40 ligand or some cytokines induced CD80 expression on B cells (15-19). Some reports showed that stimulation of CD80 and CD86 on B cells using anti-CD80 and 86 antibodies stimulated cell proliferation, immunoglobulin class switching and germinal center formation (20-23). It was reported that CD80 increased pro-apoptotic molecules and decreased anti-apoptotic molecules, but CD86 promoted activation of LPS-stimulated B cell (24).
In this study, we investigated increases of CD80 and CD86 expression during transformation of B cell after EBV infection. We aimed to examine the cell proliferation, the morphological change, the alteration of adhesion molecules and its signal pathway on EBV-transformed B cells after stimulation of CD80 and CD86 using anti-CD80 and CD86 anti-bodies.
MATERIALS AND METHODS
Preparation of EBV-infectious culture supernatant
EBV infection was induced using the cell-free supernatant containing virus particles. The super-natant was prepared from an EBV-transformed B95-8 marmoset cell line. Cells were grown in an RPMI-1640 medium (HyClone, Logan, UT, USA) supplemented with penicillin, streptomycin, and 10% fetal bovine serum (HyClone) at 37℃ in 5% CO2. The culture supernatant was harvested, centrifuged (1,000 rpm for 10 minutes) and filtered using a 0.2 µm pore-sized filter to remove cell debris.
Generation of EBV-transformed B cells
Peripheral blood mononuclear cells (PBMCs) were obtained from the blood of 5 healthy volunteers who gave informed consent for this study by Ficoll-Paque (Amersham Bioscience, Uppsala, Sweden) gradient centrifugation. B cells were purified from PBMCs using a MACS B cell-negative depletion kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Purified B cells (4×106/ml) were added to EBV stock supernatant in a culture flask, and after 2 hours of incubation at 37℃, an RPMI-1640 culture medium (Hyclone) and 1 µg/ml of cyclosporine A (Sigma-Aldrich, St. Louis, MO, USA) were added to deplete remaining T cells. The cultures were incubated for 2-4 weeks and observed every day until clumps of EBV-infected B cells were visible and the medium turned yellow. Phenotype of transforming cells was monitored using flowcytometer (FACSCalibur, BD Bioscience, San Diego, CA, USA) and confocal microscope (510 META, Carl Zeiss, Jena, Germany).
Cross-linking of CD80 and CD86 on EBV-transformed B cells
EBV-transformed cells (5×105 cells/well, 200 ul) were incubated with anti-CD80, anti-CD86 or isotype control (MOPC21, Sigma-Aldrich) for 24 hour at 37℃. Cells were harvested in a 1.5-ml tube and washed twice in cold PBS. Cells were used for following experiments. To determine optimal conditions, experiments were performed at variable concentrations (0.3124, 0.625, 1.25, 2.5 and 5 ug/ml). To inhibit accumulation of ROS, cells were pretreated with NAC (10 mM, an antioxidant; Sigma Aldrich) for 1 hour.
Detection of cell proliferation and apoptosis
To examine cell proliferation, alamar blue (Serotec, Raleigh, NC, USA) assay was performed at 72 hours after CD80 and CD86 stimulation. Alamar blue dye was added (10% by volume) to each well, and the relative fluorescence (530 nM excitation, 590 nM, 620 nM emission) was detected 7 hours later using a EL-800 fluorometer (Bio-Tek, Winnoski, VT, USA). Experiments were performed in triplicate, and the relative fluorescence value represented the mean fluorescence value determined for each culture. Reduction ratio was calculated by manufacturer's protocol. To detect apoptosis, Cells were incubated anti-CD80, anti-CD86 or MOPC21, and then harvested, washed twice with PBS, and resuspended in 100 ul of 1×binding buffer (10 mM HEPES/NaOH, pH 7.4; 140 mM NaCl; 2.5 mM CaCl2). 2 ul of FITC-conjugated Annexin V (BD Bioscience) was added and cells were incubated at room temperature for 15 minutes in the dark with gentle vortexing. Finally, cells were washed out and 400 ul of 1× binding buffer was added to each tube and cells were analyzed using FACSCalibur flow cytometer (BD Bioscience).
Measurement of ROS levels
The ROS level was detected using 2',7'-dichlorofluorescin diacetate (DCFH-DA), a fluorescence probe, which could be converted to highly fluorescent dichlorofluorescein (DCF) in the presence of intracellular ROS. EBV-transformed B cells were pretreated with 10 µM DCFH-DA (Molecular probes) for 30 minutes. Cells were then stimulated with anti-CD80, anti-CD86 antibody or MOPC. Cells were harvested, and then ROS levels determined as a FITC level using a FACSCalibur flow cytometer (BD Bioscience).
Reverse transcription-polymerase chain reaction
Total RNA was isolated using an RNAeasy mini kit (QIAGEN, Hilden, Germany). RNA was transcribed into cDNA using oligo (dT) primers (Bioneer, Daejeon, Korea) and reverse transcriptase. PCR amplification was performed using specific primer sets (Bioneer) for CD54 (forward, 5'-TGG TAG CAG CCG CAG TCA TA; backward, 5'-CTC CTT CCT CTT GGC TTA GT, 250-bp), RhoA (forward, 5'-CTC ATA GTC TTC AGC AAG GAC CAG TT; backward, 5'-ATC ATT CCG AAG ATC CTT CTT CTT, 309-bp), and Ras (forward, 5'-GAC GGA ATA TAA GCT GGT GG; backward, 5'-TAA CTA CCC CTC TGC ACG GA, 151-bp). For the control group, a specific primer set for beta-actin (forward, 5'-ATC CAC GAA ACT ACC TTC AA; backward, 5'-ATC CAC ACG GAG TAC TTG C, 200-bp) was used. PCR was performed using AccuPower PCR premix (Bioneer). PCR products were analyzed by 1% agarose gel electrophoresis and visualized with ethidium bromide under UV light using the LAS-3000 (Fujifilm, Tokyo, Japan). Data were analyzed using ImagaJ software (NIH).
Western blot analysis
After stimulation, EBV-transformed B cells were pelleted and lysed in RIPA buffer (Elpis biotech, Daejeon, Korea) with protease and phosphatase inhibitor cocktail (Sigma-aldirch). Protein concentrations were determined by BCA protein assay kit (Promega, Madison, WI, USA). The proteins (10 µg/sample) were immediately heated with sample buffer for 5 minutes at 100℃, were subjected to SDS-PAGE on gel containing 10% (wt/vol) acrylamide under reducing conditions. Separated proteins were transferred to PVDF membranes (Millipore, Bedford, MA, USA), and then the membranes were blocked with 5% skim milk. Primary antibodies (1:200~1:1,000) were incubated at room temperature for 1 hour and wash out 3 times with TBS-T buffer (0.05% tween-20). HRP-conjugated secondary antibodies were further incubated for 1 hour. Chemiluminescence was detected using an ECL-kit (Enhanced ChemiLuminescence; Amersham) and LAS-3,000 (Fujifilm). The following primary antibodies were used: anti-CD54, FAK, p-FAK, JNK, p-JNK, RhoA, beta-actin from Cell signaling (Beverly, MA, USA), anti-c-jun and p-c-jun from Santa Cruz Biotechnology.
RESULTS
Proliferation and apoptosis of EBV-transformed B cells after CD80 and CD86 stimulation
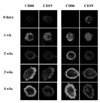
Freshly isolated B cells did not express both CD80 and CD86 on surface (Fig. 1), but intracellular staining revealed that CD86 was abundant in B cell cytosol (data not shown). After 1 week of EBV infection, intracellular and surface expression of CD80 and surface expression of CD86 were increased. At 4 week which is known as a stable period of EBV-transformation, phenotype of most cells was CD19+CD80+CD86+ determining by flow cytometric analysis (data not shown). To examine cellular response after CD80 and CD86 stimulation, EBV-transformed B cells (4 weeks) were incubated with different concentrations of anti-CD80, anti-CD86, or isotype control (IgG1, MOPC21). Stimulation of CD80 or CD86 inhibited cell proliferation after 72 hours incubation with 0.1~10 ug/ml of antibodies (Fig. 2A). In addition, apoptosis was detected at incubation with 0.3125~5 µg/ml of antibodies for 24 hours (Fig. 2B).
Reduction of CD54 expression after CD80 and CD86 stimulation
Morphological change of cell growth at early time of CD80 and CD86 stimulation was investigated using inverted microscope (Leica 351, Leica, Wetzlar, Germany). We examined that EBV-transformed B cells were forming several clumps. In contrast with isotype control, cells dispersed after 3 hours incubation with anti-CD80 or anti-CD86 antibodies (Fig. 3A). Because adhesion molecules might be involved in clump formation, we investigated whether CD54, CD44 and CD22 expression were changed after CD80 and CD86 stimulation using flow cytometric analysis. Surface CD54 expression was diminished after ligation of CD80 and CD86 (Fig. 3B), but CD44 and CD22 were not changed (data not shown). RT-PCR showed that mRNA of CD54 remarkably was decreased after 1 hour of CD80 or CD86 stimulation (Fig. 3C).
ROS generation after CD80 and CD86 stimulation
ROS (reactive oxygen species) level was investigated after CD80 and CD86 stimulation because ROS generation is associated with cellular stress. DCFH-DA was pretreated for 30 minutes before incubation with anti-CD80 and CD86 antibodies. After 4 hours stimulation, ROS was detected using flow cytometry. Ligation of CD80 or CD86 increased ROS level on EBV-transformed B cells (Fig. 4 upper panel). Both CD80 and CD86 stimulation did not increase ROS generation additively more than single stimulation. Pretreatment of NAC as a ROS accumulation inhibitor completely blocked ROS generation by CD80 and CD86 stimulation (Fig. 4 lower panel).
Ras and RhoA inactivation after CD80 and CD86 stimulation
We examined whether CD80 and CD86 stimulation modulated RhoA and Ras expression because CD54 expression is able to be regulated by Rho and Ras (25,26). RT-PCR showed that CD80 stimulation decreased transcription of RhoA and CD86 stimulation decreased transcription of Ras (Fig. 5A). Transcription of Raf-1, downstream molecules following Ras activation, was decreased after stimulation of either CD80 or CD86 (data not shown). For further study of regulation of RhoA by CD80 stimulation, phosphorylation of focal adhesion kinase (FAK), a positive regulator of RhoA was detected using immunoblot. Total RhoA and FAK proteins were not changed but phophorylated FAK and activated RhoA were diminished after stimulation of CD80 using anti-CD80 antibody (Fig. 5B). NAC pretreatment effectively recovered activation of both FAK and RhoA. Because CD86 stimulation affected only Ras transcription, MAPK (mitogen-activated protein kinase) activation following activation of Ras and Raf-1 was measured. Immunoblot showed that CD86 stimulation blocked phosphorylations of FAK, JNK, a member of MAPK family (Fig. 5C). In addition, phosphorylations of c-jun, a target molecule of JNK were decreased after CD86 stimulation.
DISCUSSION
EBV is herpes virus that infects only human, is causative agent occurring several tumors such as Burkitt's lymphoma, Hodgikin's disease, nasopharyngeal carcinoma, and gastric cancer. Abundant studies about EBV-associated tumorogenesis have been carried out, EBV-transformed B cell in vitro, lymphoblastoid cell line was usually used as a model of EBV tumorogenesis. During EBV-transformation of human primary B cell, induction of activation molecules (e.g., CD23, CD30, CD39, CD70) and adhesion molecules (e.g., CD54) were observed (27,28). These induced molecules were involved in various cell responses (e.g., apoptosis, proliferation, antibody secretion and cytokine production). We investigated EBV-transformed B cells induced CD80 and CD86, well-known costimulatory molecules (Fig. 1). Interestingly, we observed CD86 was already expressed in cytosol of resting B cells (data not shown), it was unclear why cytosolic CD86 did not express on surface. Instead, we focused cell response after stimulation of CD80 and CD86 on EBV-transformed B cells.
CD80 and CD86 are costimulatory molecules which interact with CD28 and CTLA-4 on T cells. They are expressed on antigen presenting cells such as dendritic cell, macrophage, and even B cell. CD80 and CD86 provided necessary co-activation signal into T cell through interaction with CD28. However, CD80 and CD86 for B cell activation were unnecessary. Resting B cells expressed little CD80 and did not express CD86. Activation via B cell receptor induced CD86 expression, and CD40 ligand or IL-4 stimulated CD80 expression on B cells (15-17). Thus it was not unusual for EBV-transformed B cells to induce CD80 and CD86 expression because EBV impersonated normal activation and differentiation of B cells (3). However it was not clear what viral molecules involved in CD80 and CD86 induction during EBV-transformation. To solve this, further study was required although viral late membrane proteins (LMP) were strong candidate.
When CD80 and CD86 were stimulated by monoclonal antibody, EBV-transformed B cells underwent apoptosis and growth inhibition (Fig. 2). CD80 or CD86 stimulation showed results similar to each other, but stimulation of both CD80 and CD86 on same time did not show additive or synergic effects. This supported that CD80 and CD86 shared a part of downstream signal pathway on triggering. However, another report suggested CD86 on LPS-stimulated B cells increased B cell activation in contrast with pro-apoptotic effect of CD80 (24). This discrepancy might result from different conditions of activation. Both EBV-transformed B cells and activated B cell by LPS stimulation were not physiological, several viral proteins on EBV-transformed B cells complicated this issue.
As well as apoptosis and growth inhibition, CD80 and CD86 stimulation dispersed EBV-transformed B cells (Fig. 3A). Because EBV-transformed B cell did not have ligands interacting with CD80 and CD86, we did not think that anti-CD80 and CD86 antibody might directly interfere with cell-to-cell adhesion. Interestingly, CD54 (ICAM-1) was diminished after CD80 and CD86 stimulation (Fig. 3B and C), but CD22 or CD44 did not (data not shown). CD54 is well-known adhesion molecule and its expression was increased during EBV-transformation. We did not examine whether CD54 increased during EBV-transformation was involved in clump formation or cell proliferation. However it was possible that diminishment of CD54 might be related to cellular response because reduction of CD54, cell dispersion, apoptosis and growth inhibition occurred simultaneously. Several studies that CD54 rescued germinal center B cells from apoptosis and provided proliferation signal to T cell (29-31) could support this possibility. Further molecular studies should be performed to elucidate whether CD54 was directly linked to cellular fate of EBV-transformed B cells.
Regulation of CD54 expression was much complicated [26]. Several transcription factors (e.g., NF-κB and AP-1) were involved in transcription of CD54 and many kinases (e.g., MAPK) participated in regulation of CD54. In addition, cell stress, cytokine and growth factor affected CD54 expression. In this study, we focused focal adhesion kinase (FAK) and MAPK (mitogen-activated protein kinase). FAK and MAPK were also associated with both Ras activation and RhoA activation (25). CD80 stimulation inhibited phosphorylation of FAK and activation of RhoA (Fig. 5A and B). NAC, ROS accumulation inhibitor, completely recover activation of FAK and RhoA (Fig. 5B). This results suggested that ROS as second messenger might be involved in regulation of CD54 by CD80 stimulation. In contrast with CD80, CD86 stimulation blocked Ras transcription (Fig. 5A) and phosphorylation of FAK, JNK, and c-jun (Fig. 5C), but did not affect RhoA transcription (Fig. 5A). Interestingly, NAC did not influence inhibition of JNK activation by CD86 stimulation (data not shown) although CD86 stimulation also increased ROS generation (Fig. 4). This suggested ROS generation might be related to RhoA inactivation, but its mechanism remained unclear. Although both CD80 and CD86 stimulation reduced transcription of Raf-1 as a downstream molecule of Ras (data not shown), we focused on MAPK phosphorylation following CD86 stimulation alone because CD80 stimulation was related to rather RhoA than Ras. However, it strongly suggested a possibility that not only CD86 but also CD80 stimulation reduced CD54 expression partially through MAPK pathway. Further study was necessary to solve these problems.
Taken together, we observed that CD80 and CD86 induced during EBV-transformation of B cell were able to induce apoptosis, growth inhibition, and reduction of CD54 expression. In addition, we suggested that decrease of CD54 by CD80 and CD86 regulated through inactivation of RhoA and Ras respectively. These results contribute to understanding the function of CD80 and CD86 on activated B cell or B lymphoma. Also, it may support fundamental data and the possibility of CD80 and CD86 as a target to treat EBV-associated diseases.




 XML Download
XML Download