

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) is the treatment of choice for manyhematological malignancies as well as for severe immunodeficiency. However, the main causes of patient mortality and morbidity after allo-HSCT are relapse, graft-versus-host disease (GVHD), and infections (1,2). Various complications occurring after allo-HSCT are believed to be manifestations of systemic inflammatory response syndrome (SIRS) (3). SIRS is caused by hypercytokinemia with the excess production of proinflammatory cytokines (4). It is generally recognized that crosslinking of the cytokine network plays an important role in the inflammatory processes. Dysregulation of cytokine production may initiate and perpetuate tissue damage in those who undergo allo-HSCT.
Organ impairment in SIRS is believed to occur as a sequel to vascular endothelial damage (5). Vascular endothelial growth factor (VEGF) is a dimeric glycoprotein that increases the vascular permeability and induces the proliferation and migration of endothelial cells to form new blood vessels (6,7). The role of angiogenesis in sustaining aninflammatory response has recently become evident. VEGF acts as a potent chemo-attractant for leukocytes into neovascularization site and induces endothelial cells to produce collagenase. VEGF has also been implicated in a variety of chronic inflammatory diseases (8), and is intimately linked to the cytokine network because its expression is up-regulated by a number of cytokines and growth factors. Moreover, a recent study reported an increased expression of cytochemokines, including MCP-1, IL-8, TNF-α and IL-1β, in peripheral blood monocytes treated with the placental growth factor (9), which has a remarkably similar three-dimensional structure to VEGF (10).
VEGF might be one of principal factors regulating the immune reactions associated with acute and/or chronic GVHD (11). Recently, we have demonstrated that VEGF might play a role in GVHD protection, and the early change in the serum VEGF level may be a surrogate marker for life-threatening acute GVHD (12). Through the screening of peptide libraries, a soluble arginine-rich hexapeptide sequence, anti-VEGF peptide dRK6 which could bind to VEGF and thereby block the interaction between VEGF165 and the VEGF receptor (VEGFR) has been introduced (13). Based on these data, we examined the effect of blocking VEGF in vivo after allo-HSCT using a well-established murine allo-HSCT model of human GVHD. This study demonstrated that the clinical severity and donor T cell allo-reactivity of acute GVHD were aggravated by the blockade of VEGF in early post allo-HSCT.
MATERIALS AND METHODS
Mice and reagents
Female C57BL/6 (B6, H-2b, CD45.2+), B6.Ly-5a (CD45.1+) and B6D2F1 (H-2b/d, CD45.2+) mice were purchased from Japan SLC Inc. (Shizuoka, Japan). The age of the mice ranged from 8 to 12 weeks. The mice were housed in sterilized microisolator cages and received filtered water and normal chow or autoclaved hyperchlorinated drinking water for the first 3 weeks after the allo-HSCT. The anti-VEGF peptide, dRK6 was synthesized at the Pohang University of Science and Technology in Korea, as described previously (13). The human recombinant VEGF165 was purchased from R&D Systems (Flanders, NJ, USA).
Experimental allo-HSCT and assessment of acute GVHD
The mice underwent transplantation according to standard protocol described previously (14,15). Briefly, the recipients received a single dose of 1,100 cGy total body irradiation (TBI; cesium Cs 137 [137Cs] source). T cell-depleted bone marrow (BM) cells (10×106) and 20×106 splenocytes from the respective allogeneic or syngeneic donors were resuspended and injected intravenously into the recipient animals on day 0. Depletion of BM T cells in BM was performed using relevant MicroBeads and MACS system (Miltenyi Biotec, Bergisch Gladbach, Germany), as a instruction of manual. The CD45.1+ B6Ly-5a mice were used as donors for the donor cell expansion or engraftment experiments. The recipient B6D2F1 underwent transplantation with BM and splenocytes from allogeneic B6 or syngeneic B6D2F1 donors, respectively. Each experiment was conducted on 3 or 6 animals per group. The survival was monitored daily, and the body weights and GVHD clinical scores of the recipients were measured weekly. The degree of systemic acute GVHD was assessed usinga scoring system that incorporates 5 clinical parameters: weight loss, posture (hunching), activity, fur texture, and skin integrity. This scoring system is more accurate than just weight loss alone, as described previously (16). At the time of analysis, the mice from coded cages were evaluated and graded from 0 to 2 for each of the criteria. A clinical index was subsequently generated by adding the scores of the 5 criteria (maximum index, 10).
Anti-VEGF peptide, dRK6 treatment
To determine the in vivo effect of VEGF blockade on the severity of acute GVHD, syngeneic or recipient mice were injected subcutaneously with the dRK6 peptides or control diluent. The recipient mice were injected with 50 µg of the dRK6, dissolved in phosphate buffered saline (PBS, Gibco, Grand Island, NY), and control with PBS alone. It has been previously shown that serum levels of the inflammatory cytokines associated with the severity of GVHD after major histocompatibility complex (MHC) class I- and II- disparate allo-HSCT (B6, H-2b→B6D2F1, H-2b/d) with lethal irradiation peaked on day +7 and decreased thereafter until on day +14 (17). Because VEGF may be associated with inflammatory reactions in early post allo-HSCT, the dRK6 was injected subcutaneously into HSCT recipients for 14 days from day 0 to day +14 every other day (total 7 injections) to determine if it influences the severity of acute GVHD, whereas control mice received identical injections of the diluent. The dose of dRK6 was chosen for its ability to modulate an inflammatory disease, collagen-induced arthritis (CIA), in another model (18).
Histologic examinations
At day +28, mice were killed for histological analysis. Formalin preserved skin, liver and intestine were embedded in paraffin, cut into 5-µm-thick sections, and stained with hematoxylin and eosin for histologic examination. Slides were coded without reference to prior treatment and examined in a blinded fashion by a pathologist. A semi-quantitative scoring system was used to assess the following abnormalities known to be associated with GVHD. The skin was scored for atrophy (0, 1, 2 and 3) and lymphocyte infiltration (0, 1 and 2). The liver was scored for lymphocyte infiltration at the portal area (0, 1, 2, 3 and 4). The intestine was scored for number of crypt apoptotic cells per 10 gland area (0, 0.5, 1, 2, 3, 4 and 5). The scoring system denoted 0 as<2, 0.5 as 2~5, 1 as<30%, 2 as 30~70%, 3 as>70%, 4 as moderate apoptosis in all glands, and 5 as severe apoptosis in all glands (19).
Flow cytometric analysis
All the antibodies including phycoerythrin (PE)-conjugated anti-CD3 monoclonal antibody (mAb), fluorescein isothiocyanate (FITC)-conjugated anti-CD4 mAb, FITC-conjugated anti-CD8 mAb and APC-conjugated anti-CD45.1 mAb were purchased from BD Pharmingen (San Diego, CA, USA). The procedure was performed as described elsewhere (19). Briefly, the cells were first incubated with mAb 2.4G2 for 15 minutes at 4℃ and then with the relevant FITC- or PE-conjugated mAb for 30 minutes at 4℃. Finally, the cells were washed twice with PBS/0.2% bovine serum albumin and fixed with PBS/1% paraformaldehyde. Flow cytometry was performed using a FACSVantage SE cell sorter (BD Biosciences, San Diego, CA, USA). Double staining for CD45.1 and CD3+ (CD4+ or CD8+) was used to evaluate the expansion of donor T cells after allo-HSCT.
Cytokine enzyme-linked immunosorbent assay (ELISA)
The amount of the cytokines (IFN-γ, TNF-α and VEGF) in the sera of recipient mice was measured by ELISA using a commercially available kit (R&D Systems, Minneapolis, MN, USA). The samples and standards were run in duplicate. The absorbance at 450 nm was measured using a microplate spectrophotometer, Benchmark Plus (Bio-Rad, Richmond, CA, USA).
RESULTS
Administration of dRK6 aggravated the severity of acute GVHD after experimental allo-HSCT
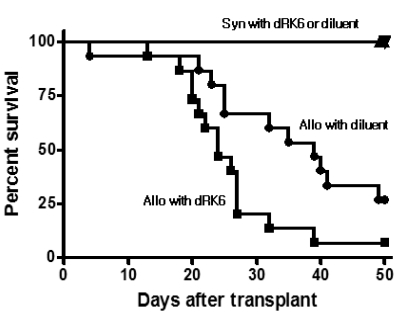
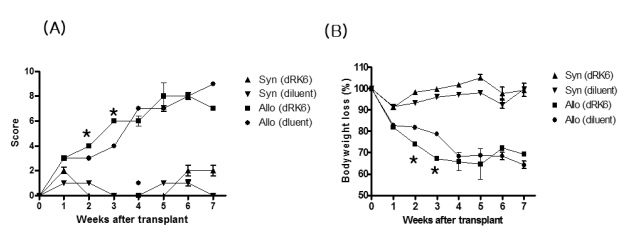
As shown in Fig. 1, injection of dRK6 after the B6→6D2F1 allo-HSCT accelerated the GVHD mortality. Allo-HSCT recipients injected dRK6 exhibited mortality more rapidly than controls with 10% of animals surviving by day +50, while the control allogeneic group mice exhibited 30% survival at the end of 50-day observation period (p=0.012). Animals that underwent syngeneic B6D2F1→B6D2F1 (syn-) HSCT showed 100% survival irrespective of the dRK6 injections, which ruled out any nonspecific toxicities of the conditioning and the dRK6 peptide injection. Fig. 2A shows that clinical GVHD in the allogeneic recipients receiving dRK6 was more severe than in control mice (p<0.05). As expected, scores for syn-HSCT recipients gradually returned to baseline by week +2. The body weight loss of recipients of each group which also correlates with the severity of acute GVHD. Consistent with the increased acute GVHD-related mortality and the clinical GVHD scores, the dRK6 injection caused significantly severe weight loss compared with the controls (p<0.05) (Fig. 2B).
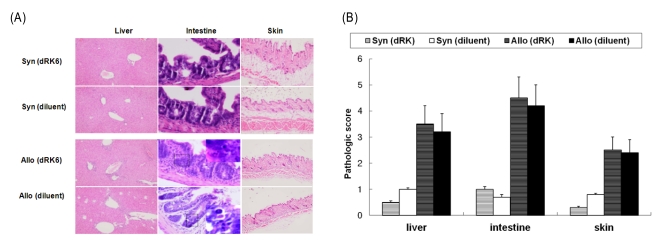
Fig. 3A shows that the allogeneic recipients had significantly more histopathologic tissue damage on day +28 compared with the syngeneic recipients. We observed that heavy lymphocyte infiltration at the hepatic portal areas as well as intestinal epithelium apoptosis in allogeneic recipients, but a few damage was found in the liver of the syngeneic recipients. In addition, we observed epidermal hyperplasia and lymphocyte infiltration in thedermis in the skin of allogeneic animals compared to syngeneic recipients. Recipients of the dRK6 injections, however, differed little from the controls in allogeneic or syngeneic recipients, respectively (Fig. 3B). Thus, the injection of dRK6 in this allo-HSCT model was associated with increased severity of acute GVHD, as determined by survival and clinical parametersbut not by the pathologic grading. Histologic examination demonstrated that this murine allo-HSCT model was adequately established to observe the occurrence of acute GVHD, which are similar to human GVHD.
Effect of dRK6 on donor T-cell expansion after allo-HSCT
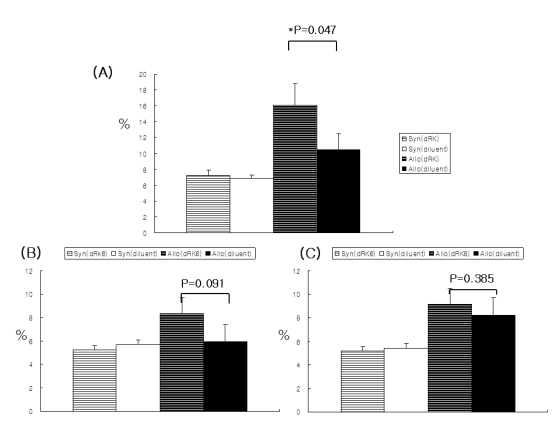
All the recipient animals displayed complete donor hematopoietic chimerism, as determined by FACS analysis (data not shown). The administration of dRK6 significantly enhanced donor CD3+ T-cell expansion in the recipient spleens on day +7 (16.1±2.7% vs 10.5±2.0%, Fig. 4A; p=0.047) in this lethally irradiated murine allo-HSCT model of B6 (H-2b)→6D2F1 (H-2b/d). As shown in Fig. 4B and 4C, dRK6 injection more clearly enhanced the expansion of donor CD4+ T cells (8.3±1.4% vs 5.9±1.5%; p=0.091) compared to that of donor CD8+ T-cell subsets. Donor CD8+ T-cell expansion by the administration of either the dRK6 or the PBS was similar (9.2±1.4% vs 8.2±2.4%; p=0.385).
Serum VEGF concentrations after allo-HSCT
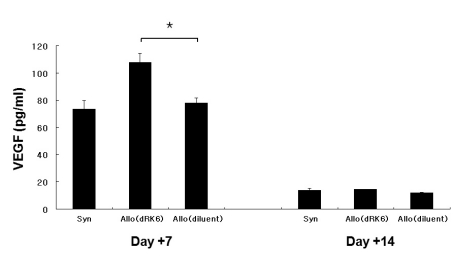
The circulating VEGF concentrations on day +7 were significantly elevated in recipients of dRK6 than in the controls (108±6.5 pg/ml vs 78±3.6 pg/ml, p=0.005) whereas VEGF levels at later time (on day +14) were similar between groups (14±1.0 pg/ml vs 12±0.5 pg/ml, p>0.05) (Fig. 5A). Both TNF-α and IFN-γ serum levels were not significantly higher at day +7 and +14 in allogeneic animals injected with dRK6 compared with the control animals (data not shown).
DISCUSSION
It was previously demonstrated that vascular endothelial damage and hypercytokinemia are involved in acute GVHD, which means that VEGF may also play a major role in modulating the graft-versus-host reactions among the many manifestations of SIRS (20-23). However, the mechanism by which VEGF functions in acute GVHD remains controversial. Lunn et al.(11) reported that serum VEGF levels were significantly raised in patients with GVHD compared with those who did not develop GVHD. However, Min et al.(12) demonstrated that there was a significant correlation between a low VEGF level and the occurrence of severe acute GVHD including grade III-IV. In the current study, we tested whether the dRK6 peptide treatment affected acute GVHD and explored the mechanisms of VEGF on the development of acute GVHD using a murine allo-HSCT model. Our results demonstrate that the high circulating levels of VEGFand magnified donor T-cell expansion caused by the blockade of the interaction between VEGF and VEGFR following the injections of the anti-VEGF peptide, dRK6, early after transplant are associated with the aggravation of acute GVHD mortality after murine allo-HSCT. Although dRK6 injection do not directly increase the production of pro-inflammatory cytokines such as TNF-α and IFN-γ, which have been importantly implicated in acute GVHD (24,25), the increased VEGF concentrations early after allo-HSCT suggest that VEGF may play an important role in the occurrence of acute GVHD.
Our results are partly in line with the previous report that high circulating levels of VEGF were associated with the attenuated severity of acute GVHD leading to a lower non-relapse mortality in human data (12). In these experiments, serum VEGF levels on day +7 were significantly higher after administration of dRK6, but those on day +14 became decreased and similar irrespective of a large dose of dRK6 injections (Fig. 5). These results indicate that consumption of VEGF in GVHD target tissues may be augmented in the allogeneic recipients that experienced more severe acute GVHD caused by administration of dRK6. Moreover, the local expressions of hepatic VEGF protein between day +7 and day +14 have a tendency to increase in the allogeneic recipients with no respect to the VEGF blockade (data not shown). We postulate that circulating VEGF in early posttransplant was excessively used during the occurrence of acute GVHD, indicating that a condition of low VEGF expression after allo-HSCT may be associated with aggravation of the severity of acute GVHD.
It has been shown that the dRK6 strongly inhibited VEGF-induced angiogenesis with retarded growth of tumor cells (13) and the severity of CIA (18). In our study, it is remarkable that, on the contrary, the administration of dRK6 in a well-established B6 (H-2b)→B6D2F1 (H-2b/d) acute GVHD model was associated with the higher mortality and morbidity. Indeed, acute GVHD mediated mainly by CD4+T cells can be inhibited by the early administration of pro-inflammatory cytokines such as IFN-γ, IL-2, IL-12, and IL-18 (19,26-28). The reason why the severity of acute GVHD is attenuated is attributed to the fact that the pro-inflammatory cytokines cause Fas-dependent apoptosis of donor CD4+ T cells rather than donor CD8+ T cells. In this allo-HSCT model in which acute GVHD is also mainly mediated by CD4+ T cells, the administration of dRK6 inhibited the biological effect of VEGF which may inhibit CD4+ T-cell mediated GVHD. Our data indicated that the donor CD4+ and CD8+ T-cell subsets showed a trend toward differential expansion caused by the dRK6 injection. Allogeneic recipients with dRK6 administration had a tendency to show a greater donor CD4+ T-cell expansionin spleen compared to those injected the control diluent, but donor CD8+T-cell expansion by the administration of either the dRK6 or the PBS was similar (Fig. 4B and C). Therefore, caution must be exercised before applying cytokine manipulation in general to the clinical situation in which both T-cell subsets play an important role in mediating GVHD.
One of the earliest physiological responses to a tissue injury or infection is an increase in vascular permeability and blood flow to the affected area, which initiated by regional vasodilation followed by enhanced angiogenesis to facilitate the wound healing process (29). Cytokine signaling also plays an important role in the angiogenic response to wound repair and infection. VEGF is an endothelial cell-specific mitogen with an ability to promote angiogenesis (30). As wound maturation progress, additional angiogenesis is induced by increasing tissue levels of VEGF, which peak several days after injury. Anti-angiogenesis treatment with anti-VEGF peptide, dRK6 may prevent wound repair by blocking wound healing effect of VEGF in the GVHD target organs and increase the GVHD-associated morbidity and mortality.
In conclusion, the administration of dRK6 peptide after allo-HSCT aggravates the morbidity and mortality of acute GVHD by means of increased circulating VEGF production and donor T-cell expansion early after transplant. The kinetics of circulating VEGF levels earlyafter allo-HSCT showed that VEGF may have an important role in the pathophysiology of GVHD by being consumed during development of acute GVHD.
 XML Download
XML Download