

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Familial hypophosphatemic rickets (FHR) is defined as a group of disorders caused by a defect in renal phosphate transport, leading to phosphate wasting and hypophosphatemia. FHR is also characterized by the abnormal regulation of vitamin D metabolism, resulting in inappropriately normal 1, 25-dihydroxyvitamin D concentrations despite hypophosphatemia (1). X-linked hypophosphatemic rickets (XLH; MIM 307800), the most common form of FHR, is characterized by rickets and osteomalacia, lower-extremity deformities, short stature, bone pain, dental abnormalities, and abnormal vitamin D metabolism (2). Other less common forms of FHR include autosomal dominant hypophosphatemic rickets (ADHR), hereditary hypophosphatemic rickets with hypercalciuria, and tumor-induced osteomalacia.
XLH results from loss-of-function mutations in the PHEX gene (phosphate-regulating gene with homologies to endopeptidases on the X chromosome), located on Xp22.1 (3). PHEX is a member of the M13 family of type II cell-surface-membrane zinc-dependent proteases, which include neprilysin (NEP), two endothelin-converting enzymes (ECE-1 and -2), the KELL antigen, and damage-induced neuronal endopeptidase/X-converting enzyme (4). PHEX cDNA has been cloned (5) and consists of 22 exons spanning 2,247 bp of genomic sequence. Seventeen of the 22 exons are less than 130 bp long (2). PHEX and NEP share conserved genomic structures. Like NEP, PHEX includes a short N-terminal tail, a single N-terminal hydrophobic region corresponding to a transmembrane domain, a highly conserved zinc-binding domain in exons 17 and 19, and several conserved cysteine residues and amino acids that, in NEP, are involved in its catalytic activity (1).
Several studies have identified mutations in the PHEX gene in individuals with XLH. Recently, we studied the clinical and molecular characteristics of Korean patients with XLH. In this report, we describe eight different PHEX mutations identified in 15 unrelated Korean patients with hypophosphatemic rickets, including five novel mutations.
MATERIALS AND METHODS
Subjects
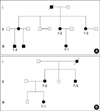
This study included 15 patients and five of their family members, aged from 20 months to 60 yr (average, 22 yr). Of the 15 patients, five had a family history of XLH, four were sporadic cases, and the other six were unknown. Diagnoses were made based on clinical, radiological, and laboratory findings by specialists at the Korea University Guro Hospital. Of the 15 patients, four were male and 11 were female. Overall, there were five male and 15 female individuals (including family members). Five patients were young children. Of the five family members evaluated, three were related to patient 1-1 (mother, maternal aunt, and cousin) and two to patient 7-1 (mother and maternal aunt) (Fig. 1).
For phenotypic analyses, the medical records and histories of the patients were reviewed retrospectively. The severity of the skeletal disease was assessed by orthopedic surgeons and was classified as mild, moderate, or severe (Table 1) (1). Osteotomies were performed in patients who complained of gait disturbance caused by either pain or fatigue. For the two patients with affected family members, the families were analyzed as a unit. They were classified as having mild disease if all members had mild disease, as having moderate disease if at least one member had moderate disease, or as having severe disease if at least one member had severe disease.
Although there are no widely accepted criteria with which to describe the severity of dental disease manifestations in patients with rickets, we simplified the assessment of dental disease severity by describing it in terms of the number of dental abscess lesions and the treatments performed for these abscesses (Table 1). The data on dental diseases were collected based on the histories of the patients.
Mutation analysis
Informed consent for DNA analysis was obtained from the patients or their parents, depending on the patient's age. Genomic DNA was extracted from the peripheral blood using the G-DEX™ II Genomic DNA Extraction Kit (Intron, Seongnam, Korea), according to the manufacturer's protocol. Screening for mutations was performed with PCR amplification and direct sequencing. All 22 exons of the PHEX gene, including at least 40 bp of the exon-intron flanking regions, were amplified by PCR. Sequencing was performed with a Dynamic™ ET Dye Terminator Kit (GE Healthcare, Buckinghamshire, U.K.) and a MegaBACE 500 Genetic Analyzer (GE Healthcare), according to the manufacturer's instructions. Base calling of the sample files was performed with Cimarron Base Caller version 3.12 software (GE Healthcare).
The PHEX genes of 50 normal female individuals were also analyzed to confirm that the sequence variations in the PHEX gene identified in this study were not polymorphisms but real pathogenic mutations. Novel mutations were defined by their absence from the Human Gene Mutation Database (http://www.uwcm.ac.uk/uwcm/mg/hgmd0.html) and from mutations previously reported in PubMed (http://www.ncbi.nlm.nih.gov/PubMed/). The functional consequences of novel splice variants were predicted with the Automated Splice Site Analyses program on the web (https://splice.cmh.edu/) (6).
Statistical analysis
The Wilcoxon rank-sum test and the two-tailed Fisher's exact test were used to calculate p values and to determine whether the differences between the phenotypes of the genotype groups were statistically significant. We used a significance level of p<0.10 because the sample size was small, in accordance with previously published analyses of small samples (1, 7).
RESULTS
In a total of 15 patients, the average ages at onset and diagnosis were 31 months and 90 months, respectively. The male: female ratio among the patients was 4:11 (Table 2, 3). Laboratory findings were available for 10 patients: seven patients with a PHEX gene mutation and three patients without a PHEX gene mutation. The mean total serum calcium and phosphorus levels for those 10 patients were 9.2±0.4 mg/dL and 2.2±0.8 mg/dL, respectively.
Eight different mutations in the PHEX gene were detected in nine patients of the 15 unrelated Korean patients (60%). Of these, c.64G>T, c.1699C>T, c.466_467insAC, c.1174-1G>A, and c.1768+5G>A were novel mutations identified in this study (Fig. 2).
Inspection of the mutations revealed that c.1363G>T (p. Glu455X) and c.1699C>T (p.Arg567X) were nonsense mutations, and that c.1601C>T (p.Pro534Leu) and c.64G>T (p. Ala22Ser) were missense mutations. One mutation, c.466_467 insAC, was an insertion that causes a frameshift leading to a downstream stop codon at amino acid 222. Three mutations, c.1174-1G>A (IVS10-1G>A), c.1768+5G>A (IVS17+5G >A), and c.1965+1G>A (IVS19+1G>A), occur at splicing acceptor/donor sites; c.1174-1G>A was detected in two unrelated Korean patients. Two mutations, c.64G>T and c.466_467insAC, are located in the N-terminal half and the other six mutations are in the C-terminal half of the PHEX protein (Fig. 3). For novel splice variants, automated splicing mutation analysis predicted that the c.1174-1G>A variation decreases the binding energy of the natural splice acceptor site to 0.5% and thus abolishes the site, and that the c.1768+5G> A variation decreases the binding energy of the natural splice site to 8.7% of its initial energy and thus weakens the original donor splice site.
The phenotypes of the 15 patients and of all available family members (including five family members of two patients) were analyzed. The average age at onset was 64 months in the group with a PHEX mutation and 17 months in the group without a PHEX mutation (p=0.049; Table 4). No significant correlation was found between the severity of the skeletal or dental disease and the mutation type (Table 5). Skeletal disease was more severe in the group with a mutation in the C-terminal half of the protein (p=0.083) (Table 5).
DISCUSSION
XLH is the most common form of FHR and results from a mutation in the PHEX gene. In our study, the mutation detection rates in the PHEX gene were 60% in familial cases and 25% in sporadic cases among patients with XLH. These are similar to the results of previous studies: 51-86% in familial cases and 22-57% in sporadic cases (1, 2, 8-10), except for one Finnish study, in which the mutation detection rate was 100% for familial cases and 93% for sporadic cases (11). The lower mutation detection rate in sporadic cases might be explained by the fact that the sporadic disease can be caused by other types of hypophosphatemic rickets, such as ADHR (1). Thus, patients without a PHEX gene mutation should be screened for mutations in fibroblast growth factor 23 (4, 12). Because only the 22 exons were screened for mutations in our study, mutations in the promoter, introns, 5'-untranslated region, 3'-untranslated region, and target sequences of miRNAs might have been overlooked (10, 13).
The c.1601C>T mutation has been observed in many studies (1, 2, 8-10, 14). This region may be prone to mutation, as suggested by Dixon et al. (9). Pro534 is encoded by one of 47 CpGs in the PHEX gene (15). The p.Pro534Leu mutation seems to affect the local hydrophobicity of the gene product, because leucine is aliphatic (2), and the substituted proline is conserved in ECE-1 and the KELL antigen (8). The c.64G> T mutation, a new mutation detected in this study, occurs at the site of the putative transmembrane domain. No mutation was identified in the putative transmembrane domain in a previous study (9).
The marked difference in the average age at onset between the groups with and without a PHEX mutation may be the result of the small sample size and the late age at onset of one family member, subject 7-2 in Table 2. Severe phenotypes usually begin to manifest at an earlier age than do mild phenotypes. However, early mean age of onset and disease severity were not correlated in this study. Furthermore, the difference in the average age at onset between the groups with and without a PHEX gene mutation was not statistically significant in a previous study (10). A larger sample size is required to confirm these data.
The number of mutations in the C-terminal half of PHEX was 3.5 times greater than the number in the N-terminal half in this study, and 1.7 times greater than that reported by Filisetti et al. (15). Patients with a mutation in the C-terminal half of the protein had more severe skeletal disease in this study. The N-terminal region contains the cytoplasmic domain, transmembrane domain, and five conserved cysteine residues, whereas the C-terminal region contains two zinc-binding motifs in exons 17 and 19, five conserved cysteine residues, and the catalytic site (2, 5, 15, 16). No agreement has yet been reached regarding the correlation between the disease phenotypes and PHEX mutation locations, including those in this study (1, 8, 17). Holm et al. (1) identified a trend between truncating mutations and more severe skeletal disease in a familial group (p=0.072). However, Cho et al. (10) reported no correlation between phenotype and mutation type, consistent with the results of this study.
The gene dosage effect was also analyzed in the nine patients with PHEX mutations, but no significant correlation was found (data not shown). Because the sample was small, we did not divide the group into prepubertal and postpubertal age groups. However, Holm et al. (1) reported a trend towards an association between male sex and more severe dental disease. Because XLH is inherited as an X-linked dominant trait, females should usually be less severely affected than males. However, no gene dosage effect was observed in this study or in a previous study (10), although our sample was too small to allow any conclusion to be drawn. Previous studies have indicated that the PHEX gene is subject to random inactivation, but it has also been reported that some alleles escape inactivation (18, 19).
Hypophosphatemic rickets often exhibit high pulp horns, large pulp chambers, and dentinal clefts. Although odontoblast function is normal, hypophosphatemia leads to dysplastic and poorly mineralized areas of the interglobular dentin (20). We classified dental diseases according to the number of dental abscess lesions and the treatments performed for these abscesses. However, no genotype-phenotype correlation was detected in comparisons of dental disease severity.
In this study, although skeletal disease tended to be more severe in the group with a mutation in the C-terminal half of PHEX, the sample size was too small to draw a definitive conclusion. Therefore, identification of the mutation in the PHEX gene may have a limited prognostic value in patients with XLH. Other factors may exist that determine the phenotypes of patients with XLH.







 XML Download
XML Download