

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Centronuclear myopathy (CNM) is a congenital myopathy with the characteristic morphology of centrally located nuclei in the vast majority of muscle fibers. After the disease was first reported in 1966 by Spiro et al. as myotubular myopathy (1), three distinctive inheritance patterns in CNM have been discovered, i.e., a severe neonatal onset with X-linked recessive type (XRCNM), a less severe infantile or childhood onset with autosomal recessive type (ARCNM), and a milder late childhood or adult onset with autosomal dominant type (ADCNM) (2). To date, six cases have been reported in Korea (3-7). XRCNM is a relatively uniform disorder clinically characterized by severe hypotonia, rapidly progressive muscle weakness, and respiratory failure at birth often leading to early mortality. This form is also called as X-linked myotubular myopathy. However, unlike XRCNM, the clinical features of autosomal dominant CNM (ADCNM) are quite variable among reported families.
We describe unusual manifestations of ADCNM with preferential involvement of the distal lower extremities, especially in the gastrocnemius and soleus muscles, with a brief review of clinical features of previously reported ADCNM.
CASE REPORT
Case 1
A 49-yr-old woman presented with slowly progressive weakness of both legs, which had developed at the age of forty. Initially, she felt her gait waddling slightly, and she had difficulty in climbing stairs and standing on her toes. Weakness was dominant in the distal part of both lower extremities, which did not affect daily activities. She was a product of a full-term spontaneous vaginal delivery, without perinatal or developmental problems. She had no history of medical illness or drug use, including steroids and herb medicines. She had two dughters. One was an 18-yr-old girl, who developed similar symptoms at the age of fifteen (see case 2), and the other, a 15-yr-old girl, was not affected. Her 70-yr-old mother had the same gait abnormality and progressive weakness and wasting of both calf muscles, which occurred in late-fifties without impairment of daily activities. Other family members did not have a history of neuromuscular diseases. Neurological examinations revealed a mild symmetrical weakness of the distal lower extremities. Muscle powers of the ankle dorsiflexors and plantar flexors were Medical Research Council (MRC) grade 5- and 4, respectively. Muscle-strength testing was otherwise normal. There was marked atrophy of both calf muscles (Fig. 1). Deep tendon reflexes were hypoactive on both upper and lower extremities without pathological reflexes. She had a mild waddling gait, and toe gait was more impaired than heel gait. There was no myotonia, fasciculation, winged scapula, or pseudohypertrophy. Examinations of cranial nerves and sensations revealed no abnormality, and her cognitive function was normal.
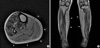
Her serum creatine kinase (CK) was slightly elevated to 304 IU/L (normal, <270 IU/L), but other routine laboratory tests, including thyroid function, were all within normal limits. Electrocardiogram, pulmonary function test, and nerve conduction studies were unremarkable. Needle electromyography showed low-amplitude, short-duration, polyphasic motor unit potentials along with fibrillations and positive sharp waves in the left vastus lateralis, tibialis anterior, and gastrocnemius muscles. There was no myotonic discharge. The magnetic resonance image of the lower extremities revealed predominant atrophy and fatty changes of bilateral gastrocnemius and soleus muscles (Fig. 2). A muscle biopsy from the right tibialis anterior showed centrally located nuclei in the vast majority of muscle fibers with marked fiber size variations. A moderate degree of endomysial fibrosis and fatty changes were also noted. NADH tetrazolium reductase staining showed type 1 fiber predominance (more than 95%) and hypotrophy, while some fibers revealed radially arranged sarcoplasmic strands (Fig. 3). Electron microscopy also showed marked fiber size variations and centrally located nuclei in myofibers frequently. All of these pathologic features suggested centronuclear myopathy.
Case 2
The 18-yr-old daughter of case 1 complained of difficulty in walking and during physical exercise from the age of 15. The neurological examinations showed a weakness in the ankle dorsiflexors and plantar flexors (MRC grade 5-), impaired toe and heel gaits, mild atrophy of both calf muscles, and hypoactive tendon reflexes. Examinations of cranial nerves, sensations, and cognitive function revealed no abnormality. Nerve conduction studies were unremarkable. Needle electromyography revealed low-amplitude, short-duration, polyphasic motor unit potentials along with fibrillations and positive sharp waves in the right tibialis anterior and gastrocnemius muscles. The patient refused further laboratory tests.
DISCUSSION
Our patients presented with muscle weakness and atrophy localized mainly in the posterior compartment of the distal lower extremities. Ophthalmoplegia, ptosis, or facial weakness was not observed. Similar clinical features were noticed in her mother and daughter. The disease progression appeared to be very slow. At presentation, these patients were believed to have a form of distal myopathy, a group of genetically and pathologically heterogenous diseases characterized by the pattern of weakness, age of onset, pattern of inheritance, and muscle pathology (8). The pattern of muscle weakness and atrophy in our patients closely resembled Miyoshi myopathy. However, the calf muscle weakness and atrophy typically starts in late teens in Miyoshi myopathy (9), and the inheritance pattern is also different. Histopathological findings in our patients clearly showed central nuclei in most fibers, radial arrangement of sarcoplasmic strands around these nuclei, and the predominance of and hypotrophy (smallness) of type 1 fibers. All these morphological features are typical findings of CNM (10).
The clinical features of ADCNM were reviewed in detail by Wallgren-Pettersson et al. (11). The usual manifestations were slowly progressive generalized muscle weakness, often predominantly proximal. Some patients, however, showed additional distal involvement. Occasionally, diffuse hypertrophy (12), calf pseudohypertrophy (13), facial weakness (14), ptosis, or ophthalmoplegia (11) was reported.
In a recent study of three ADCNM families (15), two subgroups were found: two families showed diffuse muscle weakness of late onset, whereas the third family presented a diffuse muscle hypertrophy. In that study, it was concluded that no clinical subgroups could be identified based on muscle weakness distribution, which was diffuse in most patients with an occasional proximal or distal predominance. An ADCNM family with a clinical picture simulating facioscapulohumoral syndrome has also been described (16). In our cases, however, the unique feature of the family members was weakness and atrophy of plantar flexors of both legs without ptosis or ophthalmoparesis, and this pattern was identical in the three affected family members. The younger age of onset in subsequent generations suggested an anticipation phenomenon in our family. With regard to predominant distal weakness of legs, one report mentioned familial cases with weakness and atrophy localized mainly in distal arms and legs. However, these cases showed hypertrophy of calves (13). Recently, Fischer et al. reported on the clinical and muscle imaging findings of ADCNM patients with dynamin 2 (DNM2) mutations (17). In those patients, muscle computerized tomography revealed early and predominant involvement of distal muscles, later affection of the posterior part of the thigh and gluteus minimus muscles. Considering the similarity in clinical presentations between those cases and ours, DNM2 mutations may also be responsible in our cases.
Our report demonstrates the expanding clinical heterogeneity of autosomal dominant centronuclear myopathy and emphasizes the importance of muscle pathology in the definitive diagnosis of cases with hereditary myopathies. Further analysis will be necessary to clarify the responsible gene defect in our cases.


 XML Download
XML Download