

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Although the natural history of diabetic nephropathy for patients with type 2 diabetes is not well delineated, five stages characterize the progression of this disease. During early stage type 2 diabetes, renal hyperperfusion and increased glomerular filtration develop and are followed by stages of microalbuminuria that progress to overt nephropathy and renal failure. Multiple biochemical mechanisms have been proposed to explain the progression of diabetic nephropathy, including hyperglycemia-induced increased polyol pathway flux, increased accumulation of advanced glycation end products, the activation of protein kinase C, and increased hexosamine pathway flux (1). In addition to hyperglycemia-induced biochemical factors, hemodynamic abnormalities including systemic and glomerular hypertension play a critical role in the progression of diabetic nephropathy (2).
There is a hypothesis that salt-sensitive hypertension, including hypertension in diabetic nephropathy, results from abnormally enhanced tubuloglomerular feedback and impaired pressure natriuresis (3). One of the important mechanisms underlying abnormal renal sodium handling involves the altered expressions of sodium transporters in renal tubules (4). Recently, a variety of specific antibodies have been produced against renal sodium transporter proteins (5-8). Thus, it is now possible to follow changes in the expression of sodium transporters in response to systemic physiologic disturbances by using immunoblotting and immunohistochemistry. Many investigators have described the altered expression of renal sodium transporters in streptozotocin-induced diabetes mellitus rat models or animal models which have diabetic nephropathy with early renal insufficiency (9-11).
In the present study, Otsuka Long-Evans Tokushima Fatty (OLETF) rats were used as an animal model of type 2 diabetes mellitus, and Long-Evans Tokushima Otsuka (LETO) rats were used as their diabetes-resistant counterparts. OLETF rats developed from Long-Evans rats showed higher blood glucose levels than the LETO control rats after 18 weeks of age (12). Observed sequential histologic changes in OLETF rats were glomerular enlargement and mesangial expansion after 22 weeks of age, mesangial sclerosis and exudative lesions after 36 weeks of age, and total glomerular obsolescence of almost 14% of glomeruli at 46 weeks of age (13). To study long-term alteration of expression of renal sodium transporters in the diabetic nephropathy of type 2 diabetes, studies were performed in 40 week-old animals. We undertook this study to determine how these sodium transporters are regulated during the progression of diabetic nephropathy in type 2 diabetes.
MATERIALS AND METHODS
Animals
Experiments were performed using male OLETF rats as the diabetic group (n=6) and male LETO rats as the control group (n=6). These animals were kindly donated by Otsuka Pharmaceuticals (Tokushima, Japan). All animal procedures were approved by the animal ethics review committee of the Clinical Research Institute of Seoul National University Hospital. Rats were housed individually and had free access to standard rat chow and tap water. Physiologic data were collected at 12, 25, 30, and 40 weeks of age, and animals were sacrificed at 40 weeks of age.
Physiologic measurement
Three days before physiologic data collection, rats were individually placed in metabolic cages for adaptation. Body weights were monitored. Systolic blood pressure (SBP) was measured 3 times at tail arteries, and average values were used (Noninvasive Blood Pressure system; Harvard Apparatus, Holliston, MA, U.S.A.). Twenty-four hour urine was collected and the amounts were measured. We analyzed urine electrolyte and creatinine (Cr) levels using an ion-selective method (System E4A; Beckman Coulter Inc., Fullerton, CA, U.S.A.) and urine protein concentrations by spectrophotometry (Hitachi 7170 Automatic Analyzer; Hitachi Medical Co., Tokyo, Japan). Urine osmolality was measured using a cryoscopic osmometer (Osmomat 030-D-M, Gonotec, Berlin, Germany).
At 40 weeks, rats were anesthetized with an intraperitoneal (i.p.) injection of ethyl carbamate (Choohgwae Pharma Co., Seoul, Korea), and laparotomies were conducted. Blood was collected from the aortas, and glucose, Cr, and Na levels were measured (Hitachi 747 Autoanalyzer; Hitachi Medical Co.). Glycosylated hemoglobin (HbA1c) was measured using a high performance liquid chromatograph (Bio-Rad VARIANT turbo-II hemoglobin testing system; Bio-Rad Laboratories Inc., Hercules, CA, U.S.A.). Creatinine clearance (Ccr) and fractional excretion of sodium (FENa) were calculated using measured values.
Semiquantitative immunoblotting
We evaluated the abundances of major renal sodium transporters from whole-kidney homogenates by immunoblotting. After blood collection, right kidneys were rapidly removed. Whole kidneys were homogenized in 10 mL of an ice-cold isolation solution (250 mM sucrose, 10 mM triethanolamine, 1 µg/mL leupeptin, and 1 mg/mL phenylmethylsulfonyl fluoride (pH 7.6) at 15,000 rpm with 3 strokes for 15 sec with a tissue homogenizer (PowerGun 125; Fisher Scientific, Pittsburgh, PA, U.S.A.). After homogenization, total protein concentrations were measured by bicinchoninic acid protein assays (BCA Reagent Kit; Sigma, St Louis, MO, U.S.A.) and adjusted to reach the same protein concentration (2.5 µg/µL) with isolation solution. Samples were stabilized by adding 1 vol of 5×Laemmli sample buffer to 4 vols sample and heating at 60℃ for 15 min. To confirm equal protein loading, "loading gels" were made for each sample set to allow fine adjustment of loading amounts. Five micrograms of protein from each sample were loaded into individual lanes and electrophoresed on 12% polyacrylamide-SDS minigels using a Mini-PROTEAN II electrophoresis apparatus (Bio-Rad Laboratories Inc.) and then stained with Coomassie blue dye. Three selected bands from these gels were scanned (SLB Mylmager SL-6 Digital Imaging System; UVP Inc., Upland, CA, U.S.A.) to determine the densities and the relative amounts of protein loaded per lane. Finally, protein concentrations were corrected to reflect these measurements. The same amount of protein was loaded in each lane of 8% or 10% polyacrylamide-SDS minigels and electrophoresed. The proteins were then transferred electrophoretically to nitrocellulose membranes (Bio-Rad Laboratories Inc.). After blocking with 5% skim milk in PBS-T (80 mM Ha2HPO4, 20 mM NaH2PO4, 100 mM NaCl, and 0.1% Tween-20, [pH7.5]) for 1 hr, the membranes were probed overnight at 4℃ with primary antibodies and then incubated with secondary donkey anti-rabbit horseradish peroxidase conjugated antibody (31458; Pierce, Rockford, IL, U.S.A.). Sites of antigen-antibody reaction were viewed using enhanced chemiluminescence substrate (LumiGLO Reserve Chemiluminesecnce Substrate Kit; KPL, Gaithersburg, MD, U.S.A.) and exposure to radiography film (Hyperfilm; Amersham Pharmacia Biotech, U.K.). To facilitate semiquantitative immunoblotting, we normalized the band densities by dividing by mean values of corresponding control group values; calculated values were expressed as percentages.
Immunohistochemistry
Each left kidney was perfused by retrograde perfusion via the abdominal aorta by inserting a perfusion needle into the abdominal aorta and dissecting the inferior vena cava to establish an outlet. Blood was washed from the kidneys using cold 1% phosphate-buffered saline (PBS). After completing this perfusion, the kidneys were removed, sliced into 5-mm-thick pieces, and immersed in 4% paraformaldehyde solution (P6148 in PBS, pH 7.4; Sigma) overnight at 4℃. After embedding in paraffin, kidneys were sectioned at 5 µm thickness using a microtome (RM 2145; Leica Instruments, Nussloch, Germany).
Paraffin sections were then de-paraffinized with 100% xylene and rehydrated with graded ethanol. To retrieve antigens, sections were boiled in a preheated target retrieval solution (0.01 M, pH 6.0, S2031; DakoCytomation, Carpinteria, CA, U.S.A.) 3 times for 10 min using a microwave. Sections were then treated with Dual Endogenous Enzyme Block (K4065; DakoCytomation) for 10 min to eliminate endogenous peroxidase activity, washed with distilled water and PBS, incubated overnight at 4℃ with primary antibodies diluted in PBS, and washed with buffer solution. Labeled Polymer-HRP (DakoCytomation) was then applied to the sections, and they were then incubated for 40 min at room temperature. After the slides were rinsed with substrate buffer, 3,3'-diaminobenzidine (DAB) chromogen solution (DakoCytomation) was applied, and the slides were incubated for 10 min. This reaction was stopped with distilled water. Sections were counterstained with hematoxylin, and the slides were washed with tap water, dehydrated, and mounted.
Primary antibodies
In the present study, we used previously characterized polyclonal antibodies, namely, affinity-purified polyclonal antibodies against sodium hydrogen exchanger 3 (NHE3) (5), bumetanide sensitive NaK2Cl co-transporter (NKCC2) (6), thiazide-sensitive NaCl co-transporter (NCC) (7), and the α-, β-, and γ-subunits of the epithelial sodium channel (ENaC) (8).
Statistics
Values were presented as means±standard errors. Quantitative comparisons between groups were determined using the Mann-Whitney U-test. Statistical analyses were conducted using SPSS software (SPSS Inc., Chicago, IL, U.S.A.), and p values of <0.05 were considered statistically significant.
RESULTS
Physiologic parameters
The mean serum glucose level of OLETF rats was elevated to 235±25 mg/dL at 25 weeks and hyperglycemia continued to 40 weeks of age (30th week: 260±42 mg/dL, 40th week: 275±30 mg/dL, Fig. 1A). However, LETO rats did not develop hyperglycemia (25th week: 126±10 mg/dL, 30th week: 122±14 mg/dL, 40th week: 140±9 mg/dL, Fig. 1A). The HbA1c values of OLETF rats were also higher than those of LETO rats at 40 weeks of age (LETO vs. OLETF: 3.7±0.0 vs. 6.3±0.2%, p<0.05, Table 1). Body weights of OLETF rats were greater than those of LETO rats between 12 and 30 weeks of age (12th week: 322±5 vs. 393±6 g, p<0.05; 25th week: 435±13 vs. 547±23 g, p<0.05; 30th week: 522±17 vs. 647±6 g, p<0.05, Fig. 1B), but were not different at 40 weeks of age (557±20 vs. 509±25 g, Fig. 1B). SBP values of OLETF rats were higher than those of LETO rats at 25 and 30 weeks of age (25th week: 138±6 vs. 175±6 mmHg, p<0.05; 30th week: 152±4 vs. 193±4 mmHg, p<0.05, Fig. 1C), but were not significantly different at 40 weeks of age (127±2 vs. 140±5 mmHg, Fig. 1C). As diabetic nephropathy progressed, urine volumes and protein excretion also increased. Twenty-four hour urine volumes per kg of body weight increased after 25 weeks of age in OLETF rats, and this difference between the 2 groups increased progressively until 40 weeks of age (40th week: 15±3 vs. 106±33 mL/day/kg, p<0.05, Fig. 1D). The mean urine protein/Cr ratio of OLETF rats was 10 times higher than that of LETO rats at 40 weeks of age (1.0±0.1 vs. 11.8±2.5 mg/mg, p<0.05, Table 1). However, serum Cr levels were lower in OLETF rats (0.75±0.02 vs. 0.65±0.02 mg/dL, p<0.05, Table 1) and Ccr levels were higher at 40 weeks of age (1.6±0.2 vs. 3.3±0.2 mL/min/kg Bwt, p<0.05, Table 1). FENa values of OLETF rats showed a tendency to be lower than those of LETO rats at 30 and 40 weeks of age; however, the difference was not statistically significant (Fig. 1E). The amounts of daily excretion of sodium tended to be insignificantly higher than those of LETO rats at 30 and 40 weeks of age (Fig. 1F, Table 1).
Immunoblotting
The abundance of NHE3 at 40 weeks of age was not different between the two groups (LETO vs. OLETF: 100±8 vs. 103±17%, Fig. 2A). The abundances of NKCC2 (100±27 vs. 108±18%, Fig. 2B) and NCC (100±8 vs. 77±8%, Fig. 2C) were not significantly altered according to the progression of diabetic nephropathy. However, the subunits of ENaC showed heterogeneous responses to the disease progression. The abundance of the ENaC α-subunit was not significantly different between the two groups at 40 weeks of age (100±37 vs. 49±5%, Fig. 3A). However, the abundance of the ENaC β-subunit was significantly increased (100±13 vs. 171±20%, Fig. 3B) in OLETF rats, and the abundance of the ENaC γ-subunit was significantly reduced (100±18 vs. 32±9%, Fig. 3C) in OLETF rats at 40 weeks of age.
Immunohistochemistry
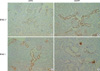
Compatible with immunoblotting results, the ENaC β-subunit showed markedly increased immunoreactivity in the renal cortex of OLETF rats compared with those of LETO rats at 40 weeks of age (Fig. 4A, B), whereas the ENaC γ-subunit showed reduced immunoreactivity in the renal cortex in these rats at this time (Fig. 4C, D).
DISCUSSION
Blood glucose levels in OLETF rats increased beyond the diagnostic criteria of diabetes at 25 weeks of age, and hyperglycemia continued to study termination at 40 weeks of age. In addition, the mean HbA1c level in OLETF rats was higher than that in LETO rats at 40 weeks of age. These findings are consistent with previous reports that OLETF rats show elevated glucose levels at 18 weeks of age (12). After the development of hyperglycemia, SBP values were elevated in OLETF rats. Moreover, 24-hr urine volumes, urine protein/Cr ratios, and Ccr levels were higher in OLETF rats than in LETO rats at 40 weeks of age. Although the differences were not statistically significant, the amounts of daily excretion of sodium tended to be higher than those of LETO rats at 30 and 40 weeks of age. The findings resulted from increased urine volumes of OLETF rats. Indeed, FENa values of OLETF rats showed a tendency to be lower than those of LETO rats at 30 and 40 weeks of age. According to our physiologic data and a previous report about histologic changes in OLETF rats (13), the stage of diabetic nephropathy at 40 weeks of age in this model reflected overt nephropathy before the development of renal failure.
In the present study, we analyzed changes in the abundance of the major sodium transporters in renal tubule, as these may be responsible for hemodynamic changes that occurred in-line with the progression of diabetic nephropathy. Our results show that the sodium transporters expressed in the proximal tubule, the thick ascending limb of loop of Henle, and the distal convoluted tubule were not different in age-matched OLETF and LETO rats. However, ENaC subunit expression in the second part of the distal convoluted tubule, connecting tubule, and collecting duct showed heterogeneous changes in OLETF rats.
In diabetic obese Zucker rats, another type 2 diabetic animal model, the abundances of NHE3 in the cortex and outer medulla and of NKCC2 in the outer medulla were lower than in those of nondiabetic lean Zucker rats at 6 months of age (11). However, in the present study, the abundances of NHE3 and NKCC2 in OLETF and LETO rats were similar at 40 weeks of age. We believe that these differences are due to different stages of diabetic nephropathy in the two animal models. In the present study, the mean Ccr per kg of body weight for OLETF rats was higher than in LETO rats. However, in diabetic obese Zucker rats, Ccr per kg of body weight was lower than in lean Zucker rats. Therefore, the reduced abundances of NHE3 and NKCC2 of diabetic obese Zucker rats probably reflected glomerulotubular balance because GFR was beginning to fall at this time. In an early study of younger Zucker rats, which showed insulin resistance but which were not diabetic, no differences were found between lean and obese rats with respect to the abundances of NHE3 and NKCC2 (14). These findings suggest that the proximal tubule and thick ascending limb transporter expressions are not changed during early diabetic nephropathy before renal function deteriorates.
The distal portion of the renal tubule, including the distal convoluted tubule and the collecting duct, plays an important role in the fine tuning of sodium reabsorption. NCC in the distal convoluted tubule is under the control of aldosterone, which up-regulates NCC expression (7). However, in the present study, the abundance of NCC was not changed in OLETF vs. LETO rats. This indicates that NCC expression in OLETF rats is not only modulated by aldosterone but also influenced by other factors.
In the present study, GFR was increased in OLETF rats vs. LETO rats, which might have increased the distal delivery of tubular fluid. Abdallah et al. reported that increased sodium delivery to the distal tubule by high dose furosemide infusion increased NCC protein abundance (15). However, the abundance of NCC was not changed by a low dose furosemide infusion (16). These findings suggest that the amount of tubular fluid delivered to the distal tubule influences the expression of NCC. In addition to aldosterone and the distal delivery of tubular fluid, other mechanisms related to diabetic nephropathy progression also regulate the abundance of NCC. In younger Zucker rats, the abundance of NCC was increased in obese rats (14). However, consistent with the findings in our OLETF rats, this increased expression of NCC was not found in older diabetic Zucker rats (11). Song et al. also recently reported that chronic insulin infusion and drinking of glucose did not change the abundance of NCC (17). Therefore, It is difficult to understand that NCC expression is related with the progression diabetic nephropathy.
The ENaC is a protein that is required for the reabsorption of sodium in the connection tubule through the collecting duct. ENaC is tetramer and is composed of α, β, and γ-subunits (18). Moreover, the physiologic activity and the abundance of each subunit of ENaC is under the control of aldosterone. Masilamani et al. reported that dietary NaCl restriction and aldosterone infusion markedly increase the abundance of the ENaC α-subunit in rat kidneys without increasing those of β- and γ-subunits (8). In their study, aldosterone was found to induce a shift in the molecular weight of the ENaC γ-subunit from 85 kDa to 70 kDa. However, in the present study, the abundance of the ENaC α-subunit was not different in OLETF and LETO rats, despite an increase in the abundance of the ENaC β-subunit and a decrease in that of the ENaC γ-subunit. The results of our study demonstrate that changes in the abundance of ENaC subunits are not consistent with the effect of aldosterone on ENaC expression.
Although the regulation of ENaC is primarily under the control of aldosterone, other hormones, such as insulin (19) and vasopressin (20), may also play a role in this process. In addition to regulation by many hormones, changes in the abundance of ENaC subunits were non-coordinated to the same stimulation (8).
The up-regulation of the ENaC β-subunit abundance also was observed in younger and non-diabetic obese Zucker rats of 2 and 4 months of age, respectively (14), and the absolute abundance of the ENaC β-subunit was significantly increased in diabetic Zucker rats after correcting for the renal hypertrophy (11). Therefore, an increase in the abundance of the ENaC β-subunit is probably not a result of hyperinsulinemia or hyperglycemia, but rather may be related to either hypertension or insulin resistance. The mechanism of reduced ENaC γ-subunit abundance needs to be investigated in the future. Different methods of regulation of these ENaC subunits appear to be related to many other factors that are associated with the development and progression of diabetic nephropathy. The physiologic meaning of these different methods should be elucidated.
Hypertension is one of the most important risk factors for the progression of diabetic nephropathy (21). An increase in sodium and water retention is required for the development of most forms of hypertension. However, pressure natriuresis, which protects the kidneys from sodium retention and hypertension, may not be fully functional in salt sensitive-hypertensive states, such as diabetic nephropathy. One of the mechanisms whereby the kidney regulates sodium reabsorption is via altered expression of renal tubular sodium transporters, especially ENaC (4, 22). Because of the lack of a downstream sodium transport system beyond ENaC, sodium reabsorption by ENaC in the collecting duct becomes the final renal adjustment of sodium balance. The results of many studies indicate that an increase in sodium transport by ENaC may be a common and important component of salt-sensitive hypertension (23, 24). Therefore, the altered expression of ENaC subunits in OLETF rats may be an important mechanism of abnormal salt handling and hypertension which are largely responsible for the progression of diabetic nephropathy.
In conclusion, the abundance of the ENaC β-subunit was founded to be increased and that of ENaC γ-subunit to be decreased in overt proteinuric stage diabetic nephropathy in the present murine type 2 diabetic model. We suggest that these changes are related to an altered sodium balance, which might be associated with hemodynamic changes, and that this is responsible for the progression of diabetic nephropathy.




 XML Download
XML Download