

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Bipolar disorder (BD) is unique among psychiatric conditions in that its symptoms swing between two opposite mood states: mania and depression.1 Almost 70 years ago John Cade found out that lithium had beneficial effects in his patients suffering from "mania, dementia praecox and melancholia."2 The favorable outcome with lithium treatment in patients with major mental illnesses was a breakthrough in that era. Since that discovery, however, no pharmacological agent for the specific treatment of BD has been ascertained, and for all intents and purposes the management of this condition is with psychotherapeutic drugs developed for other indications.3 In the past few decades, more and more refined investigational techniques have been employed to uncover the pathophysiology of BD. As a result, several important discoveries have been made; the translational nature of the research gives rise to the expectation that new insights will lead to more effective treatments for patients with BD and that the fruits of scientific knowhow will be passed on from the bench to the bedside.
BD is a complex medical condition whose etiology involves genetic and epigenetic factors acting alongside environmental stresses in causing expression of the disease.4 This diathesis is currently viewed as a multisystem ailment that not only affects brain functioning but also results in physical comorbidities like cardiovascular disease, diabetes mellitus, disorders of immunity, and endocrine dysfunction. Genome-wide studies have failed to detect any single gene to account for the incidence of BD, fostering the prevailing assumption that it is a polygenic condition.5 The involved genes interact with life stresses to cause disruption in biological and homeostatic mechanisms. Research efforts in recent years have disclosed that dysregula tion of vital biochemical pathways acts in an orchestrated manner in the pathogenesis of BD. There is disruption of glucocorticoid signaling, immune-inflammatory imbalance, increased oxidative stress, abnormalities of tryptophan metabolism, and derangement of phospholipid turnover with shifts in the 1-carbon cycle of methionine and homocysteine. 6 These identified mechanistic pathways offer opportunities for the development of novel and state-of-the-art therapeutic agents, which hold the promise of opening fresh avenues in the treatment of BD.
Life stresses acting on predisposed individuals have an enduring effect on the neural substrate, causing rewiring of the nervous system with increased sensitization and proneness to recurrent affective episodes.7 There is mounting evidence that manic and major depressive exacerbations have a neurotoxic effect, damaging the neurons as well as the glial elements in the brain.8 The results of preclinical and human studies consistently show accumulating organ damage both in the center and in the periphery with illness progression. The neuroprogressive nature of BD is clinically manifested as increased frequency and severity of episodes, greater suicidal risk, and cognitive and functional impairment.9 In the final stages of the disorder there is no illness remission, persistence of inter-episode subthreshold affective symptoms, and eventual loss of autonomy.10 The course of BD is malignant in many cases; currently available medications fail to control the disease manifestations, with very high rates of polypharmacy, soaring frequency of treatment-emergent adverse effects, and meager compliance from the patients.11 Considering that BD is a prevalent condition, the application of less than optimal treatment strategies and consequent illness progression place a huge burden on the individual patient, his or her family, and society as a whole. In view of these concerns, it is vitally important to cultivate better understanding of the disease mechanisms so that adequate cures are made available to the numerous people afflicted by this intractable illness. The purpose of this was review to clarify the key interacting pathophysiological mechanisms that drive the disease process in bipolar spectrum disorders and to uncover potential new treatment targets for these and related mood disorders.
SEARCH STRATEGY
In August 2015, the PubMed electronic database was searched for literature on the neurobiology of mood disorders, with particular reference to BD. The investigative approach was broadly based to include articles on animal models with extrapolative value for human disease. During the appraisal of the literature, special emphasis was placed on the translational significance of the research work. This policy was employed with the specific intent to uncover the pathophysiology of bipolar spectrum disorders while at same time identifying novel therapeutic targets for the treatment of these conditions. The aim of conducting a wide-ranging evaluation of the extant body of work was driven by the understanding that the management of BD is particularly challenging considering the heterogeneity, developmental trajectory, and neuroprogressive nature of the disease. An attempt was made to highlight original concepts in the neurobiology of BD that were both innovative and had heuristic value. During the preparation of the article, only studies conducted in the last 10 years were included. Several combinations of search terms were used; some examples are "mood disorders and biology," "bipolar disorder and HPA axis," "bipolar disorder and cytokines," "bipolar disorder and circadian," "mood disorders and circadian rhythm," "mood disorders and inflammation," "bipolar disorder and oxidative stress," and "bipolar disorder and neurobiology." Investigative work on animals and humans as well as review articles were assessed; articles that were considered to be especially pertinent were read in full and their reference lists were also consulted. Finally, the data were integrated in a narrative review in a concise and coherent style.
HYPOTHALAMIC-PITUITARY-ADRENAL AXIS DYSFUNCTION
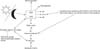
Early in the trajectory of BD, episodes occur secondary to stress but there is blighted psychobiological resilience and defective coping that increase vulnerability to recurrent affective exacerbations with illness advancement. 12 This impairment is principally provoked by the hypothalamic-pituitary-adrenal (HPA) axis, which does not function properly in patients with BD.13 Patients with BD have a hyperactive HPA axis, high levels of systemic cortisol, and nonsuppression of its circulating levels in the dexamethasone suppression test or the dexamethasone/corticotrophin-releasing hormone (DEX/CRF) test.14 Furthermore, subjects with increased susceptibility, such as first-degree relatives, have also been shown to have increased baseline cortisol levels and aberrant responses to the DEX/CRF test.15 From this perspective, HPA axis irregularities seem to be a genetic attribute endowing vulnerability to mood disorders. The glucocorticoid receptor (GR) is the most important factor in the formulation of the cortisol response; it binds to the hormone in the cytosol and shuttles it to the nucleus where it functions as a transcription factor. The action of the GR is dependent on a hefty molecular complex consisting of several chaperone proteins and cofactors, including FK506 binding protein 51 (FKBP51).16 In vitro experiments in humans reveal that overexpression of FKBP51 reduces hormone binding affinity and nuclear translocation of GR, whereas high levels of this chaperone cause GR insensitivity and raised peripheral levels of cortisol in nonhuman primates.17 Intriguingly, this hormone acting via an intracellular ultra-short negative feedback loop for GR activity can stimulate the expression of FKBP51.18
There is a known familial contribution to the neurobiology of BD, and it is probable that most of the cortisol-GR-related mechanisms alluded to above are a sign of the putative genetic underpinning. In this regard, research has shown that the hereditary component in BD largely acts through gene-environment associations. In essence, biological and psychosocial stressors reprogram gene activity by altering epigenetic modifications, thus escalating the risk for the disease in predisposed people, as well as meddling with the illness trajectory in those with the expressed phenotype. Among the purported epigenetic modifications, alterations in DNA methylation have been repeatedly shown in bipolar patients.19 Significantly, in murine models, prolonged exposure to glucocorticoids is known to bring about changes in DNA methylation at the FKBP5 gene. In human studies, such alterations have been noted in the Fkbp5 gene in patients with a stressor-related comorbid condition of BD, namely, post-traumatic stress disorder. As such, FKBP5 methylation may be one of paths through which the HPA system acting in response to stress malfunctions in BD pathophysiology.20 Given that the mechanisms of HPA axis dysregulation are incompletely known at present, as is its role in dictating the risk of the disease in vulnerable subjects, current work is beginning to unravel the molecular targets of illness development and progression in BD.21 The crucial function of GR in the actions of cortisol is depicted schematically in Fig. 1.
IMMUNOLOGICAL FACTORS
1. Immune-inflammatory imbalance
In bipolar patients, major mood episodes of either polarity result in an inflammatory response that has been convincingly shown in several studies. This is evident as an increase in the levels of proinflammatory cytokines (PIC) and C-reactive protein in the peripheral blood.22 The PIC include most importantly interleukin 1β (IL-1β), IL-6, and tumor necrosis factor-alpha (TNF-α). Treatment with mood stabilizers and resolution of acute affective exacerbations has been shown to normalize the levels of some PIC like IL-6 but not TNF-α, whereas a chronic, low-level inflammatory state persists even in euthymic patients.23 This points to the fact that immune-inflammatory dysregulation is fundamental to the pathophysiological processes in BD.24
It is suggested that unrelieved strain in BD activates the HPA axis with increased secretion of cortisol; in addition, there is stimulation of the sympatho-adrenal medullary axis with increased circulating levels of epinephrine and norepinephrine. These stress hormones acting by virtue of their membrane and cytosolic receptors affect the transcription of genes encoding cytokines via decreased inhibition of the canonical nuclear factor-kappa B (NF-κB) and the noncanonical inflammatory signal transduction pathways like activator-protein 1 (AP-1), Janus kinasesignal transducer and activator of transcription (JAK-STAT) factors, and mitogen-activated protein kinases (MAPK).25 The cells of the innate immune system (for example, monocytes, macrophages, and T-lymphocytes) secrete PIC, chemokines, and cell adhesion molecules that diffuse to the brain and trigger microglia, causing neuroinflammation and further activating the HPA axis.26 The unrelenting secretion of stress hormones leads to a constant low-grade inflammatory milieu in the body that is considered to be liable for the neuroprogression of the bipolar diathesis and also predisposes to cardiovascular and metabolic abnormalities often encountered in these patients. 27 Additionally, with repeated mood episodes and advancement of BD, there is a shift of the immune response from T-helper 2 (TH2) cells, which mainly secrete anti-inflammatory mediators like IL-10, to TH1 cells, which produce PIC (for example, TNF-α).28 Several case-control studies, mainly having cross-sectional designs, have revealed dysregulation of the immune-inflammatory response in BD. Table 1 summarizes recently published work in this area.
2. IL-6 trans-signaling in mood disorders
IL-6 is a ubiquitous inflammatory cytokine performing diverse biological actions that vary from regeneration and repair of cellular elements to augmenting the response to injury in various types of tissue damage.35 Several studies have documented an increase in peripheral circulating levels of IL-6 during acute mood episodes of either polarity.36 Because the brain is no longer considered privileged from the effects of peripheral inflammatory mediators, IL-6 gains access to this organ from the circulation. Upon stimulation, microglia and astrocytes also secrete IL-6 locally.37 However, the receptor IL-6R, which binds to this cytokine in nanomolar concentrations, is expressed by a few cell types only (including some leukocytes and hepatocytes). The complex of IL-6 and IL-6R binds with a second glycoprotein, gp 130, that subsequently dimerizes and kicks off intracellular signaling via the JAK/STAT pathways.38 Unlike IL-6R, gp 130 is expressed on all cells; however, the latter alone has no affinity for the cytokine and cells lacking IL-6R cannot respond to this mediator. Of note, a soluble form of IL-6R (sIL-6R) consisting of the extracellular portion of the receptor was detected in humans in plasma and other body fluids. sIL-6R is generated by partial proteolysis of the membrane-bound receptor by "a disintegrin and metalloprotease" (ADAM17).39 The sIL-6R interacts with IL-6 with similar binding properties as the membrane-bound IL-6R. IL-6 trans-signaling is the path by which gp 130–expressing cells, even in the absence of membrane-bound IL-6R, can be stimulated by the complex of IL-6 and sIL-6R.40 Because of this phenomenon, the inflammatory process can affect every organ system in the body, but under the steady state condition, uncontrolled inflammation is kept in check and physiological homeostasis is maintained.41
In the course of inflammation, proteolysis of the IL-6R from neutrophils by virtue of ADAM17 leads to the activation of endothelial cells which do not express IL-6R on their membranes and are therefore insensitive to the cytokine. Priming of the endothelial cells by the IL-6/sIL-6R complex leads to the secretion of the mononuclear cell attracting cytokine MCP-1. Thereby, the shedding of the IL-6R acts as a measure of preliminary injury as shown by the number of neutrophils involved, since these cells are the first to arrive at the site of damage.42 Furthermore, experimental models in mice have determined that classic IL-6 signaling has a regenerative and anti-apoptotic role during inflammation, for example, in the cecal puncture and ligation sepsis paradigm during which the animals are subjected to extreme stress. In contrast, IL-6 trans-signaling reflects the proinflammatory arm of the cytokine's biological activities.43 Since the proteolysis of IL-6R is mainly governed by ADAM17, it is highly likely that ADAM17 has a key function in inflammation-related phenomena (Fig. 2).44
There are therapeutic implications in this pattern, as specific agents can be developed that block the proinflammatory properties of IL-6 without stalling its anti-inflammatory actions.45 Many if not all neural cells are the target of IL-6 trans-signaling, and inhibition of this activity can be expected to have important salutary affects in neuropsychiatric disorders.46 A soluble fusion glycoprotein sgp 130Fc has been engineered from the extracellular portion of gp 130 that exclusively restrains IL-6 trans-signaling. Administration of this engineered protein is a viable treatment approach in major psychiatric conditions, including BD.47 Thus, current evidence provides a rationale for undertaking preclinical studies in mood disorders to determine the therapeutic prospects of IL-6 blocking strategies.
OXIDATIVE STRESS
1. Physiological role of free radicals
In biological terms, oxidative stress can be considered as a continuing discrepant interaction between antioxidants and prooxidants with a tilt toward the latter.48 The result of this fact is the disproportionate formation of free radicals, the reactive oxygen species (ROS). At low physiological concentrations, ROS perform important functions in the central nervous system (CNS).49 These include regulation of the destiny of neurons either through growth or programmed cell death via stimulation of the AP-1 transcription factor and the nerve growth factor pathways.50 ROS take part in critical signaling cascades such as the regulation of the membrane potential and cellular H+ fluxes, the execution of cardiovascular homeostasis and management of blood pressure through the angiotensin II receptor, and the control of the glutamatergic neurotransmission via the N-methyl-D-aspartate (NMDA) receptor.51 In addition, ROS are engaged in the neuroinflammatory response by means of priming of the microglia.52 Free radical actions may have a key function in fine-tuning the responses of neuronal cells to adverse events, either by promoting resiliency through stress-induced molecular cascades such as MAPK or by getting rid of the severely impaired cells by apoptosis.53
2. Pathophysiological mechanisms
On the flip side, the buildup of ROS is found to enhance the vulnerability of brain tissue to damage and has an important part in the pathophysiology of a number of neuropsychiatric conditions. Unequivocal proof of increased brain oxidative damage has been revealed for neurodegenerative conditions like Alzheimer's and Parkinson's diseases, cerebrovascular disorders, demyelinating diseases, and severe psychiatric ailments such as schizophrenia, major depressive disorder, and BD.54 ROS cause glutamate excitotoxicity and alter mitochondrial activity. Mitochondrial dysfunction in turn causes NMDA receptor up-regulation and further increases oxidative stress, resulting in a detrimental self-sustaining and aggravating cellular process.55 Oxidative stress products such as superoxide and hydroxyl radicals have been shown to induce cortisol resistance by impairing the GR movement from the cytosol to the nucleus. HPA axis dysregulation by ROS in turn causes a proinflammatory response with increased circulating levels of PIC.56 Unrestricted ROS activity is also responsible for increased blood-brain barrier permeability through launching of matrix metalloproteinases and subsequent degradation of tight junctions, unrepressed neuroinflammation, and enhanced apoptosis of neurons.57
3. Redox balance in the brain
The assumed pathophysiological association between oxidative stress and mood disorders may be due to the fact that the nervous system is increasingly susceptible to oxidative damage for a number of reasons. First, of all the organs, the brain has the highest consumption rate of oxygen; it is roughly 2% of body weight but uses 20% of total inspired oxygen. This fact predisposes it to greater formation of ROS during the process of mitochondrial energy metabolism. Second, the brain's lipid content is very high, and lipids act as a substrate for the ROS. Third, there is the redox potential of several neurotransmitters, for instance, dopamine. Fourth, the defense systems against free radicals are relatively inefficient. Last, the brain has a high content of metal ions, for example, iron and copper, involved in redox reactions. 58 The protective arrangement against prooxidants is composed of an enzymatic and a nonenzymatic component. Glutathione peroxidase and glutathione reductase are recognized intracellular antioxidant enzymes. The former changes peroxides and hydroxyl radicals into benign moieties, with the oxidation of reduced glutathione (GSH) into the oxidized form glutathione disulfide (GSSG), and glutathione reductase regenerates GSH from GSSG. Additionally, other enzymes such as catalase (CAT) and superoxide dismutase (SOD) also take part in the cellular resistance against oxidative stress and act along with glutathione peroxidase, thus forming the principal enzymatic defense against accumulated free radicals. Further, glutathione-S-transferase and glucose-6-phosphate dehydrogenase are important in sustaining a stable provision of metabolic products like GSH and nicotinamide adenine dinucleotide-phosphate (NADPH), which are required for proper operation of the main antioxidant enzymes.59
4. Measurement of oxidative stress
The main ROS include the superoxide radical (O2˙-), hydrogen peroxide (H2O2), and the hydroxyl radical (OH˙). Interaction between ROS and nitric oxide results in the formation of highly reactive nitrogen species, such as the peroxynitrite ions (ONOO-), which are very damaging to the basic structural molecules of the cells.60 Because ROS and reactive nitrogen species possess very short half-lives, these are hard to quantify. Therefore, it is necessary to search for and determine indirect pointers of oxidative stress; these include lipid, protein, and DNA peroxidation indicators. Whereas measurements of key enzymes involved in the neutralization of free radicals also serve as putative markers.61 Biosignatures of oxidative stress in body fluids include oxidized proteins like protein carbonyls and oxidized lipids such as 4-hydroxynonenol, thiobarbituric acid–reactive substances, malondialdehyde, and isoprostanes, which are isomers resulting from the oxidation of arachidonic acid. In addition, neuroprostanes are emerging as definitive markers of oxidative stress emanating from the CNS.62 The oxidation of DNA at the guanine residue site produces 8-hydroxydeoxyguanosine, which is associated with telomere shortening detectable in peripheral blood cells or fibroblasts.63 Telomere shortening is an indicator of accelerated aging and premature mortality owing to the increased incidence of cardiovascular and other comorbidities in major psychiatric conditions.64 There are reliable data from work on circulating markers that the brain's main antioxidants, namely, GSH, CAT, SOD, and GSH peroxidase, have changed values in bipolar subjects. In this vein, recent meta-analyses have shown increased biomarkers of oxidative stress.65 Foremost discoveries are enhanced lipid peroxidation, DNA injury, and increased amino acid nitration in bipolar patients in contrast with healthy subjects, with lipid peroxidation being the predominant finding.66
5. NADPH oxidase enzymes as a potential therapeutic target
The sole purpose of NADPH oxidase (NOX) enzymes is the generation of ROS, and their induction is purportedly a significant cause of oxidative stress in major psychiatric disorders.67 These enzymes utilize cytoplasmic NADPH and catalyze the transfer of electrons to molecular O2 to produce ROS. Acting along the transmembrane electron transport chain, NOX enzymes generate ROS in the extracellular space or the lumen of intracellular organelles. Seven NOX genes have thus far been discovered, and the best described isoform is NOX2.68 These enzymes are found in several tissues of the body, and the presence of NOX transcripts has been confirmed in total brain samples as well as in neurons, microglia, and astrocytes. In the presence of severe life stress, these are induced, causing increased production of ROS in the brain and thereby playing a role in the expression of the main psychiatric disorders.69 Animal experiments in rodents and primates with paradigms such as social isolation and establishment of communal hierarchy models have been very helpful in revealing the contribution of these enzymes to prooxidant injury and the resulting expression of different types of abnormal behavior. In the case of persistent severe life stress, the neurophysiologic impact of excessive ROS generation gives rise to continued excitatory, glutamatergic transmission leading to changes suggestive of alterations seen in psychotic spectrum disorders. These include down-regulation of the NMDA receptors, failure of the inhibitory trait of GABAergic interneurons, and reduction of hippocampal volume.70 In view of these animal experiments, the cause of oxidative stress has been shown to be the increased expression of NOX enzymes. If these findings are confirmed in humans, these enzymes might emerge as potential therapeutic targets.71 Fig. 3 illustrates this supposed model of disease development in major psychiatric disorders.
INFLAMMATION, KYNURENINES, AND MOOD DISORDERS
There is limited knowledge regarding the etiopathogenesis of affective disorders, which serves as a major obstacle in the discovery of effective preventive and curative therapies. Several converging sources of evidence point to a contribution of inflammation in the neurobiology of major depressive disorder and BD with the participation of several biological systems in the initiation and progression of these conditions.72 There is crosstalk among disturbed immune, hormonal, and metabolic systems causing aberration in the functioning of neurotransmitters and leading to the manifestations of affective disorders.73 With regard to BD, circulating levels of PIC have been shown to be increased during the course of acute episodes, returning to baseline with successful mood-stabilizing treatment.74 The strongest evidence for the contributory role of inflammation in the causation of major depressive disorder comes from the observation that cytokine immunotherapy based on chronic administration of interferon-alpha and/or IL-2 to patients with kidney cancer or melanoma with metastasis induces depressive episodes in a considerable number of patients.75 Furthermore, anti-TNFα treatment with etanercept and infliximab is emerging as a possible option in the management of resistant mood disorders, either as monotherapy or adjunctive therapy.76
PIC stimulate indoleamine 2,3-dioxygenase (IDO), which is a ubiquitous enzyme that metabolizes tryptophan along the kynurenine (KYN) pathway. The latter is an essential amino acid acquired from food; 5HT is produced from about 1% of the accessible tryptophan in the body, whereas the remaining 99% is metabolized in the hepatocytes by tryptophan 2,3-dioxygenase (TDO). TDO activity is chiefly governed by the amino acid's level itself, and therefore its biological action is usually steady and not influenced by inflammatory mediators.77 During conditions of inflammation and increased oxidative stress, IDO activation in the extrahepatic tissues shifts the metabolism of tryptophan away from the liver. KYN formed by the action of IDO is further metabolized to compounds that have neuroactive properties.78 Fig. 4 shows the main enzymes and products in the KYN pathway downstream of IDO.
In circumstances of stress, extra KYN is formed in the brain in situ, whereas its central availability is further enhanced because it can pass through the blood-brain barrier. In fact, tryptophan and KYN can cross the blood-brain barrier via a sodium-independent large neutral amino acid transporter known as System L. By contrast, the remaining KYN metabolites are exclusively formed in the brain itself because these cannot cross from the periphery into the CNS.79 In summary, under the influence of biological stimuli triggered by stress, the KYN pathway is greatly stimulated in the brain. On the basis of the facts presented above, inflammation-induced mood disorders should be amenable to treatment by the following:
a) Targeting the development of inflammation, for example, with cytokine inhibitors or cytokine signaling pathway antagonists.
This approach would certainly work best when deployed in a preemptive manner before mood disorders occur.80 Once an affective episode has come about, the neuroactive KYN pathway metabolites are formed in situ in the brain and an alternate strategy aimed at repairing the inflamed brain must be put in place.
Among the products of the KYN pathway, kynurenic acid has a putative neuroprotective role by acting as an antagonist of NMDA receptor, while it also decreases glutamate levels via inhibition of α7-nicotinic receptors. 3-Hydroxykynurenine is a free radical generator, and quinolinic acid is an NMDA receptor agonist that also exerts neurotoxic effects via oxidative damage to lipids and interruption of the blood-brain barrier. The neurotoxic activity of quinolinic acid has been known for more than 30 years. This metabolite should a priori be a therapeutic target by blocking its formation or antagonizing its excitotoxic effect on NMDA glutamatergic receptors.81 However, in the context of an inflamed brain, the microglia are activated, which results in the release of large quantities of glutamate, a process facilitated by the uptake of extracellular glutamine that is converted to glutamate by the enzyme glutaminase. Moreover, oxidative stress generated by 3-hydroxykynurenine and 3-hydroxyanthranilic acid further contributes to microglia priming. In the normal brain, the clearance of glutamate is efficient because astrocytes take up this substance and convert it to glutamine via glutamine synthetase, which is recycled back to neurons for the continued formation of glutamate. However, in conditions of excitotoxicity, the extracellular concentration of glutamate can increase up to 100-fold, overwhelming this mechanism of glutamate reprocessing.82 Such pathologically increased levels of glutamate result in atrophic changes in key moodregulating areas like the hippocampus and amygdala.83
An important mechanism for the clearance of brain extracellular glutamate is represented by the extrusion of glutamate from the brain interstitial fluid into the circulation. The brain-to-blood movement of glutamate is limited owing to the relatively high levels of plasma glutamate, so that an efficient way to substantially enhance this process is to accelerate the degradation of glutamate in the blood by administering oxaloacetate, which activates glutamic oxaloacetate transaminase. This enzyme catalyzes the transformation of glutamate into 2-ketoglutarate. Blood glutamate scavenging can be enhanced further by adding recombinant glutamic oxaloacetate transaminase to oxaloacetate. The neuroprotective effect of blood glutamate scavenging has been confirmed in a number of experimental conditions associated with high brain concentrations of glutamate, such as traumatic brain injury, epilepsy, and ischemic stroke.84
Finally, to achieve personalized treatment it is necessaryto develop suitable biomarkers for identifying thosesubjects who would most likely gain by targeting the sequenceof actions leading from inflammation to major mooddisorders. The events to be marked in this chain are thetransport of KYN into the brain; the production and actionof KYN metabolites, particularly quinolinic acid; and possiblythe increased concentrations of glutamate in thebrain interstitial fluids.85
CIRCADIAN RHYTHM ABNORMALITIES IN MOOD DISORDERS
1. Circadian markers
Chronotropic representations have added to our knowledge of the neurobiology of mood disorders, and dysfunctions in the circadian system are linked with the bipolar phenotype. The evident abnormalities include hormonal dysregulations (melatonin and cortisol fluctuations), actigraphic patterns (sleep/wake cycles), and chronotypic activity preferences (dimensional variations), which serve as circadian markers.86 These aberrations are seen not only during acute affective exacerbations but also in the euthymic periods. Most of these indicators are also found in the healthy relatives of bipolar patients, thus signifying a robust familial predisposition. Therefore, these may be considered as genetic attributes of the disorder.87 A number of circadian genes are known to be correlated with BD: several studies have shown affirmative links for CLOCK, NPAS2, ARNTL1, NR1D1, PER3, RORB, and CSNK1-epsilon. Thus, disruption of the molecular circadian clock is supposedly involved in the genetic vulnerability to BD.88The circadian hypothesis has directed the use of treatment methods such as interpersonal and social rhythms therapy and chronotherapeutics (bright light therapy, total sleep deprivation). Furthermore, this hypothesis may elucidate how mood stabilizers, particularly lithium, which inhibits the enzyme GSK3β, resynchronize the molecular clock and bring about their therapeutic effects.89
2. Social zeitgeber theory
Over the years many theories have been expounded to clarify the etiology of mood disorders. The propositions include disturbance in monoamine neurotransmission, HPA axis malfunction, immune dysregulation, decreased neurogenesis, mitochondrial dysfunction, and altered neuropeptide signaling. Almost all patients with mood disorders have major interruptions in circadian rhythms and the sleep/wake cycle. No wonder that disruption in normal sleep pattern is one of the main diagnostic criteria for these conditions. Furthermore, environmental disturbances to circadian rhythms such as shift work, trans-meridian travel, and lopsided social timetables often induce or aggravate affective exacerbations.90 It has been shown in current research work that molecular clocks exist everywhere in the brain and body, where they partake in the working of nearly all physiological functions.91 The social zeitgeber premise of mood disorders assumes that stressful life events cause alterations in the sleep/wake pattern, which then affect molecular and cellular rhythms in susceptible people, inducing affective episodes. This postulate is supported by a number of clinical studies showing an unambiguous connection between the extent of rhythm disruptions and the severity of mood episodes; also, rhythms are re-established with successful treatment.92 In essence, all present therapies for mood disorders modify or even out circadian rhythms.
3. The molecular clock
The principal circadian synchronizer is placed in the suprachiasmatic nucleus of the hypothalamus where it gets light input by a direct connection, the retinohypothalamic tract. The suprachiasmatic nucleus subsequently conveys signals via direct and indirect projections all over the brain. It also manages the timing of the discharge of several peptides and hormones, including melatonin, the sleep-promoting agent. Of note, the suprachiasmatic nucleus appears to synchronize the timing of rhythms in the periphery through changes in body temperature, which serve as a common signal to harmonize multiple organ systems to the light/dark cycle.93 The molecular clock hub consists of a sequence of transcriptional and translational periodic loops that lead to the recurrent expression of clock genes on a time scale of just over 24 hours. In the primary feedback loop, circadian locomotor output cycles kaput (CLOCK) and brain and muscle Arnt-like protein 1 (BMAL1) heterodimerize and bind to E-box-containing sequences in a number of genes including the three period genes (Per1, Per2, and Per3) and two cryptochrome genes (Cry1 and Cry2). Next in the sequence, PER and CRY proteins dimerize and translocate to the nucleus, where CRY proteins can directly stall the action of CLOCK and BMAL1. Further to this mechanism, the CLOCK and BMAL1 proteins control the expression of Rev-erbα and Rora (retinoic acid–related orphan nuclear receptors), which subsequently inhibit or trigger Bmal1 transcription, respectively, by acting via the Rev-Erb/ROR response element in the promoter.
There are a number of important catalysts that adjust the timing of the molecular clock through the biochemical processes of phosphorylation, sumoylation, etc. Casein kinases phosphorylate the PER, CRY, and BMAL1 proteins, affecting their functioning and nuclear translocation. Another enzyme, glycogen synthase kinase 3 beta (GSK3β), also phosphorylates the PER2 protein, promoting its movement into the nucleus.94
4. Circadian mechanisms in mood disorders
The action of the antidepressant agomelatine, a melatonin receptor agonist and a weak 5-HT2C receptor antagonist, causes enhancement in monoaminergic neuronal activity, pointing to a regulatory function of melatonin in monoaminergic transmission and connecting the circadian and monoamine neurotransmitter systems.95 Circadian rhythm dysregulation results in an increase in PIC, and subsequently TNF-α, INF-γ, and IL-6 alter the sleep/wake cycle and circadian gene expression via NF-κB transduction. 96 Glucocorticoids show a robust diurnal variation in rhythm and crest in level just before wakening, when melatonin falls to undetectable levels in the peripheral blood. CRY proteins acting together with the GR in a ligand-dependent manner induce rhythmic suppression of its activity. Moreover, CLOCK directly acetylates GR, leading to indifference to cortisol in the morning and increased affinity at night when acetylation is annulled.97 The immediate control of GR function by circadian genes is allegedly of great importance in modulating the reaction to persistent stress.98
There is a very high incidence of metabolic diseases in mood disorders, whereas circadian oscillations in the liver, stomach, adipose tissue, and gut are very strong.99 The peptides that play a role in metabolism and feeding comprise ghrelin, orexin, leptin, and cholecystokinin, and these demonstrate a noteworthy circadian rhythm in activity. The association between corpulence, stress, and poor sleep signifies a vicious cycle, and mood disorders can be included in this sequence for a subgroup of patients.100
The nicotinamide cofactors NADPH/NADP+ and NADH/NAD+ have lately been recognized as indispensable associates of CLOCK, NPAS2, and SIRT1, establishing an immediate connection between the redox status of the cell and circadian rhythms.101 Besides, CLOCK/BMAL1 directly controls the expression of nicotinamide phosphoribosyltransferase in a circadian manner, determining the production of NAD+ in the cell over the daily cycle.102 Brain-derived neurotrophic factor (BDNF) and its receptor tyrosine kinase B (TrkB) are known to be crucial in the effects of antidepressants and mood stabilizers, and both of these have a strong circadian rhythm in expression in the hippocampus. 103 BDNF is no longer effective as a neurotrophic agent in the nonexistence of a normal diurnal rhythm of glucocorticoids, demonstrating that the daily oscillations of cortisol control the salutary activity of psychotropic drugs on neuronal growth via BDNF/TrkB signaling in the hippocampus.104 Fig. 5 gives an overview of the links between mood disorders and the circadian system.
CONTRIBUTORY NEUROBIOLOGICAL MECHANISMS IN MAJOR MENTAL DISORDERS
There are many commonalities in the psychopathology of major depressive disorder, BD, and schizophrenia. Schizoaffective disorder is an intermediary phenotype and the retention of the "not otherwise specified" category in current classificatory systems reflects that many patients with major mental disorders do not easily fall into clear-cut diagnoses. This is demonstrated in neurobiological groundwork, and recent studies bear out this actuality. Table 2 provides an outline of this area of research.
NEUROPROGRESSION IN BIPOLAR DISORDER
BD has its usual onset in adolescence or early adulthood following a stressful event. Its course is malignant in many cases, marked by repeated affective episodes of increasing severity, development of substance use disorders, and myriad physical diseases with increased morbidity and mortality. The allostatic load model has been proposed to explain the progressive nature of BD. Table 3 recapitulates representative studies delineating the pathophysiological foundations of this framework.
IDENTIFICATION OF NEW THERAPEUTIC TARGETS
Throughout this article, an effort has been made to allude to novel therapeutic measures for BD, which is in essence a recalcitrant condition. Refractoriness to conventional mood stabilizers is the rule rather than the exception and targeting the monoamine neurotransmitter systems results in less than satisfactory results for many patients. In this regard, Table 4 presents new therapeutic targets that may open fresh avenues in the treatment of BD.
UNIFYING HYPOTHESIS OF THE NEUROBIOLOGY OF BIPOLAR DISORDER
Conceptualized as a polygenic condition, the expression of BD is secondary to a stressful event. There is evidence for HPA axis dysregulation, failure of mechanisms that mediate resiliency, and breakdown of homeostasis. Fig. 6 illustrates key biological abnormalities that act in concert and explains why so many body systems are adversely affected by the bipolar diathesis.
LIMITATIONS
FUTURE DIRECTIONS
Ongoing research on the neurobiology of BD is leading to a better understanding of the condition. Dysfunction of the molecular clockwork genes has repeatedly been shown and circadian disturbances are present not only during acute episodes but also in remission. Future research should clarify how a dysregulated internal clock precipitates affective episodes and suggest measures that could lead to the prevention and early treatment of mood disturbances, which come in innumerable flavors. A vicious cycle of immune-inflammatory imbalance, increased oxidative stress, and derangement of metabolic parameters accompanied by neurophysiological disturbances is at the foundation of this disorder. Further research should elaborate this mechanism, providing a composite view of the initiation and progression of BD. Finally, more effective and targeted therapies are likely to be discovered that are not palliative but curative in nature.
CONCLUSION
A broad assessment of the current literature was done to bring to light the underpinnings of mood disorders in general and BD in particular. These are stress-related conditions with overt expression in individuals with an underlying genetic vulnerability. Modern neuroscience is utilizing animal models and conducting human research with increasingly sophisticated methods to unravel their pathophysiology. Significant strides have been made in understanding the neurobiology of affective illnesses, and in this regard new targets and biomarkers have been identified. Diverse biological systems act in concert in perpetuating the disorders. While obstacles in research remain in the basic scientific and clinical domains, there is no doubt that a representation is emerging that is providing a consolidated view regarding the development of these intractable conditions. It is hoped that new knowledge will translate into novel therapeutic measures that have both preventive and curative value for patients with bipolar spectrum disorders.








 XML Download
XML Download