

 PDF
PDF Citation
Citation Print
Print



Introduction
Parkinson’s disease (PD) is a debilitating neurodegenerative condition characterized by the progressive loss of midbrain dopaminergic (mDA) neurons, leading to motor symptoms. Our understanding of the underlying pathological mechanisms of PD has significantly advanced, highlighting the molecular and cellular pathophysiology. While current treatments offer partial symptomatic relief, they have limitations in terms of modifying the disease’s progression, underscoring the need for alternative strategies. In the field of PD research and therapy, stem cell technology has emerged as a promising avenue. It involves differentiating patient-specific induced pluripotent stem cells (iPSCs) into mDA neurons to explore pathological mechanisms and treatment targets. Stem cell-based therapies, including the transplantation of these neurons, are being actively explored as a challenging endeavor to offer personalized regenerative approaches to PD treatment.
This article delves into the present status and future potential of stem cell-based research in addressing the multifaceted challenges of PD. We summarize efforts and their effects in directly transplanting stem cell-derived cells into patients, along with their limitations and forthcoming challenges. Additionally, we explore PD disease modeling and investigations into the pathophysiology derived from these models, extending beyond transplantation approaches. We also introduce a promising model, the optogenetics- assisted alpha-synuclein aggregation induction system (OASIS), which provides unique benefits for PD research and drug development. As we confront the complex nature of PD, the integration of stem cell technology and innovative models promises to advance our understanding of the disease’s pathology, pinpoint therapeutic strategies, and potentially slow or halt disease progression. This article offers an overview of these interesting developments in PD research and therapy.
Go to : 
Current Understanding of PD Pathological Mechanisms
PD is a complex neurodegenerative disorder characterized by the progressive loss of mDA neurons in the substantia nigra, resulting in motor symptoms such as bradykinesia, rigidity, and tremors (1, 2). Extensive research has significantly advanced our comprehension of the pathological mechanisms underlying PD. One prominent feature of PD pathology is the aggregation of the protein α-synuclein (α-syn), forming intracellular proteinaceous inclusions known as Lewy bodies (1-5). Notably, α-syn pathology is observed in both sporadic and familial PD cases and is believed to contribute to neuronal dysfunction and degeneration (6, 7). Mitochondrial dysfunction represents another critical aspect of PD pathogenesis (8, 9), with a particular impact on mDA neurons in the substantia nigra. This vulnerability is substantiated by the presence of defects in mitochondrial complex I and disturbances in mitochondrial dynamics (10). These issues culminate in increased oxidative stress, impaired energy production, and compromised cellular homeostasis within mDA neurons. Furthermore, inflammation plays a significant role in PD, as evidenced by the presence of activated microglia and elevated levels of pro-inflammatory cytokines observed in affected brain regions (11). The neuroinflammatory response further contributes to mDA neurodegeneration. Additionally, PD is associated with impaired protein degradation pathways, including the ubiquitin-proteasome system and the autophagy-lysosomal pathway (12, 13). Dysregulation of these mechanisms can result in the accumulation of misfolded proteins and impaired cellular clearance processes, ultimately contributing to neurodegeneration (Fig. 1). Genetic factors have also been identified in PD, with mutations in genes such as α-syn (SNCA), leucine-rich repeat kinase 2 (LRRK2), β-glucocerebrosidase (GBA), phosphatase and tensin homolog induced putative kinase 1 (PINK1), Parkin (PRKN/PARK2) and DJ-1 (PARK7), ATPase cation transporting 13A2 (ATP13A2), vacuolar protein sorting ortholog 35 (VPS35), phospholipase A2 group VI (PLA2G6), DNAJ6, synaptojanin 1 (SYNJ1), ubiquitin C-terminal hydrolase L1 (UCHL1), linked to familial forms of the disease (14-16). Collectively, these insights into PD pathological mechanisms provide a foundation for the development of targeted therapies aimed at preserving dopaminergic function and slowing the progression of the disease.

Go to :
Limitations of PD Treatment and the Need for Alternative Approaches
Current treatments for PD primarily target the dopaminergic pathway to alleviate motor-related symptoms. These treatments include agents like levodopa, dopamine agonists, and monoamine oxidase B (MAO-B) inhibitors (17-19). Levodopa, functioning as a dopamine precursor, is the benchmark therapeutic approach for PD, effectively mitigating motor symptoms (20). Dopamine agonists, such as pramipexole or ropinirole, directly stimulate dopamine receptors, while MAO-B inhibitors, like selegiline or rasagiline, prolong the effects of dopamine (21, 22). Although these treatments offer symptom relief and improve patients’ quality of life, they have limitations in terms of providing a cure or altering the disease progression. Moreover, long-term use of levodopa can lead to adverse effects and the emergence of motor complications, including vomiting, nausea, arrhythmia, postural hypotension, motor fluctuations, and dyskinesias (23, 24). These complications significantly affect patients’ physical capabilities and overall quality of life. Additionally, these treatments may have limited efficacy in addressing the non-motor symptoms of PD, which encompass cognitive impairments, autonomic dysfunction, and psychiatric manifestations (25, 26).
Given the intricate nature of PD pathology, it becomes imperative to explore alternative avenues, such as the development of novel treatment modalities. These may involve neuroprotective agents, gene therapies, and targeted interventions, all aimed at halting or retarding the progression of PD. This approach seeks to address the multifaceted aspects of the disease and provide more comprehensive treatment strategies.
Go to :
The Potential of Stem Cell Technology in PD Research
Stem cell technology has emerged as a prospective strategy in PD research and therapy, offering valuable tools for disease modeling and drug discovery (Fig. 2) (27). Through the differentiation of patient-specific iPSCs into mDA neurons, it enables the study of disease mechanisms and identification of therapeutic targets (28). The transplantation of stem cell-derived mDA neurons offer an innovative path to replace lost or dysfunctional neurons in PD (29). Various stem cell sources, such as embryonic stem cells (ESCs), iPSCs, and adult tissue-derived stem cells (30), have been explored for generating mDA neurons suitable for transplantation. Preclinical studies and early-phase clinical trials have yielded promising outcomes including graft survival, integration, and functional improvement in PD patients (31). These advancements have the potential to revolutionize PD treatment by providing personalized and regenerative approaches.
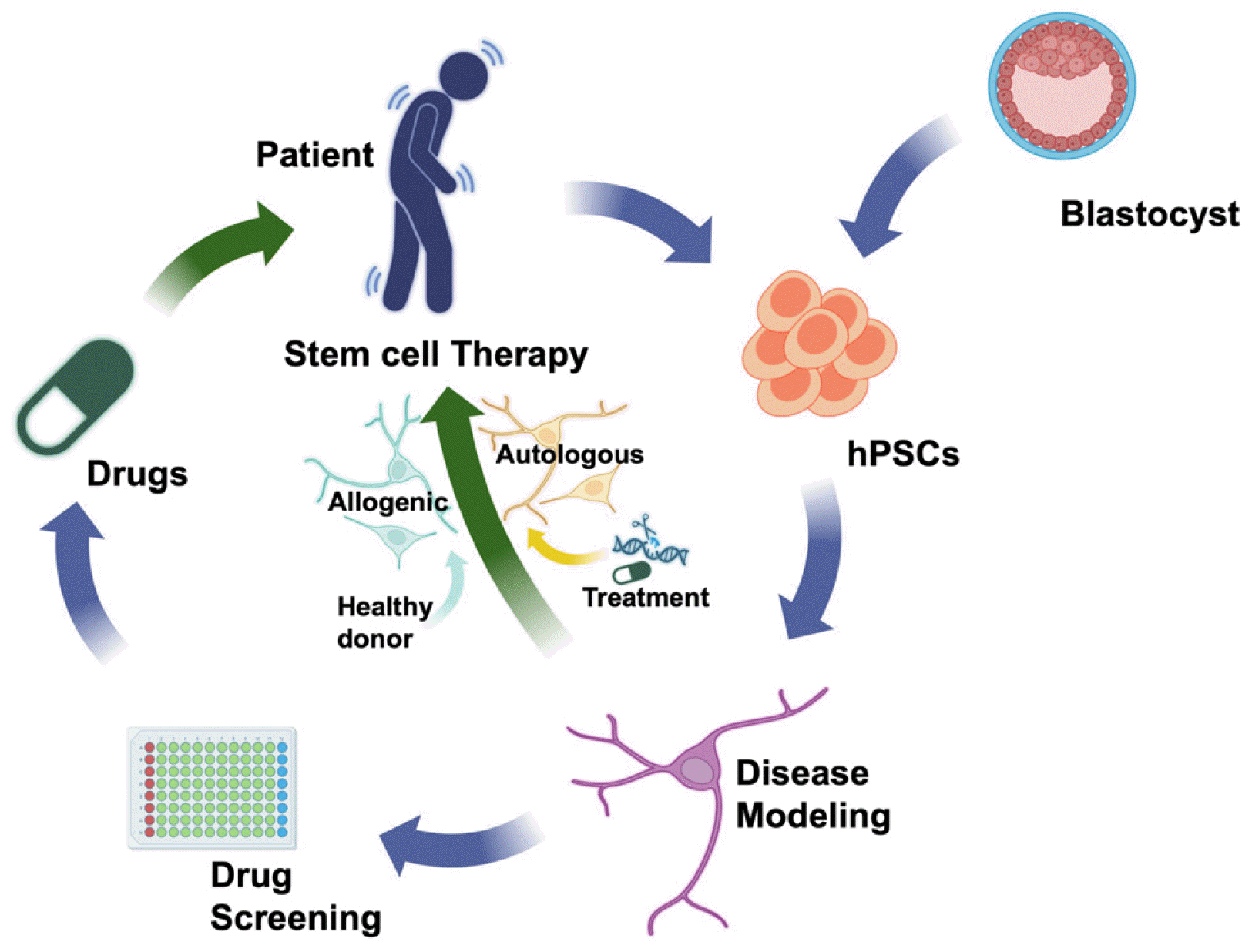
Stem cell-based therapies for PD
Stem cell-based therapies hold great promise in treating PD by addressing the potential for replacing or regenerating the depleted mDA neurons. After the discovery of adult neural stem cells (NSCs) in the human brain (32), numerous studies have explored whether neurogenesis occurs in the adult brain (33) and if restoration of mDA neurons using resident NSCs is possible in the brains of PD patients (30, 34, 35). Adult NSCs have been isolated from rodent models and human brain tissues (36), confirming their limited but verified ability to differentiate into mDA neurons (30). However, due to fate commitment issue primarily located in the anatomical subventricular zone, the efficiency of differentiation into mDA neurons from these cells was highly restricted, and the effects of animal model transplantation were modest (37). On the other hand, transplantation of fetal brain tissue into PD patients showed promising results (38, 39), but faced limitations such as tissue acquisition challenges and ethical issues. The concept of differentiating and transplanting mDA neuronal cells from human ESCs (hESCs) has emerged (40-42), leading to the investigation of two primary approaches: the transplantation of mDA neuron progenitors derived from ESCs or iPSCs, and the transplantation of NSCs (43).
ESCs and iPSCs have shown significant potential in PD therapy due to their ability to differentiate into mDA neurons suitable for transplantation (31, 44). Multiple studies have successfully differentiated ESCs and iPSCs into mDA neurons, providing a renewable source of cells for transplantation. Kriks et al. (45) generated mDA neurons from hESCs and human iPSCs (hiPSCs), demonstrating functional properties similar to native mDA neurons. Transplanting these cells into a rat PD model resulted in functional recovery and substantial improvements in motor behaviors. Kim et al. (46) developed a scalable and reproducible protocol for differentiation of hESCs and hiPSCs into mDA neurons through biphasic WNT activation. They effectively transplanted hiPSC-derived mDA neuron precursors into a primate and rat model of PD, resulting in significant enhancements in motor function and behavioral recovery (46). Schweitzer et al. (47) reported that a single patient undergoing hiPSC-derived mDA precursor transplantation exhibited stabilized or improved clinical measures of PD symptoms 18 to 24 months post-implantation. Recently, they advanced their research by co-transplanting dopamine cells with autologous regulatory T cells, effectively increasing the engraftment rate of the transplanted cells. This development signifies a shift towards not just exploring the therapeutic possibilities of cell transplantation but also seeking more efficient strategies for cell engraftment therapy (48). Several research groups have initiated clinical trials employing human PSC-derived dopamine cells, highlighting their potential for cell replacement (47, 49-52). Moreover, Piao et al. (50) and Kirkeby et al. (53) have commenced clinical trials by producing hESC-derived precursor cells of mDA neurons that meet quality, safety, and efficacy criteria for transplantation into patients, respectively. In this process, they have overcome the limitations of personalized therapy based on hiPSCs by incorporating the assistance of immunosuppressive agents (54), aiming to achieve the concept of transplantation using cryopreserved, ready-to-use cells based on hESCs.
In addition to ESC and iPSC-based transplantation, NSC-based transplantation has also shown promise for PD therapy. NSCs are multipotent cells capable of differentiating into various neural cell types, including mDA neurons. Transplantation of NSCs directly into the affected brain regions of PD patients has demonstrated positive outcomes in both preclinical and clinical studies. Roy et al. (55) successfully engrafted and differentiated NSCs into mDA neurons in a rat PD model, leading to behavioral improvements. The transplanted cells expressed mDA markers and exhibited functional integration within the host brain. In a primate model of PD, Redmond et al. (56) transplanted NSCs derived from the ventral mesencephalon, observing improved motor function, indicating the therapeutic potential of NSC-based approaches. Bachoud-Lévi et al. (57) conducted a clinical trial where NSCs were transplanted into the striatum of PD patients, demonstrating long-term survival, functional integration of the transplanted cells, along with improvements in motor symptoms and quality of life.
These studies underscore the potential of ESC, iPSC, and NSC-based therapies for PD by providing a renewable source of mDA neurons or neural progenitors capable of differentiation and functional integration. Further research and clinical trials are essential to optimize transplantation protocols, assess long-term effects, and establish the safety and efficacy of these stem cell-based therapies for PD.
Limitations of stem cell-based therapies for PD
While stem cell-based therapies offer great potential for the treatment of PD, several limitations need to be addressed at the clinical level before their extensive application. These limitations encompass concerns regarding efficacy, efficiency, potential side effects, immunological considerations, limited availability, scalability, and ethical and regulatory considerations.
Efficacy: In stem cell-based therapies for PD, efficacy is of paramount importance. Considering the diversity in genetic profiles, environmental factors, and the stages of disease progression that have resulted in variations in the effects of nearly all medications, it remains necessary to firmly establish efficacy by applying attempts to treat PD patients with dopaminergic cell transplants to a larger patient population. These efforts are still in their challenging and exploratory early stages, and, as a result, they have so far been limited to single patients or small cohorts, making it difficult to precisely evaluate the efficacy and identify influencing factors. Additionally, long-term tracking of efficacy is a vital aspect to consider. Although early clinical trials have provided promising results in terms of ameliorating motor symptoms, translating these findings into meaningful and sustained clinical benefits remains a formidable challenge. The heterogeneity in patient responses to treatment and the enduring effects of therapy necessitate further investigation. Turning to the analysis of therapeutic outcomes from the transplantation of fetal midbrain tissue in PD patients, postmortem studies revealed long-term survival of transplanted dopaminergic and serotonin neurons for over a decade (58-60). However, the presence of Lewy bodies in the transplanted neurons raises concerns about the longsuffering and stable effectiveness of stem cell-derived neuron transplants (59, 60). Conversely, another study reported no Lewy bodies observed in postmortem tissue even 14 years after cell transplantation. This divergence underscores the need for validation and optimization of stem cell-derived dopamine cell transplantation from a long-term perspective (58). Factors influencing therapeutic efficacy, including viability, functional integration, and the establishment of appropriate neural circuits within the transplanted cells, are substantial and necessitate optimization to maximize clinical advantages (61, 62).
Efficiency: Efficiency represents another significant limitation in the clinical application of stem cell-based therapies for PD. Generating a sufficient number of functional mDA neurons from stem cells in a cost-effective and scalable manner is a complex task. Crucial steps include streamlining and standardizing the manufacturing process, ensuring reproducibility and consistency of produced cells, and optimizing cell transplantation protocols. Advances in cell manufacturing technologies, including automation and quality control measures, are being explored to enhance the efficiency of stem cell-based therapies (63, 64).
Side effects: Safety concerns and potential side effects are of vital significance in the context of stem cell-based therapies. While these therapies aim to restore the function of mDA neurons, there is an inherent risk of adverse effects. One notable concern pertains to the development of graft-induced dyskinesias, characterized by abnormal involuntary movements resulting from the excessive release of dopamine by transplanted cells (65). Strategies to mitigate this risk encompass refining transplantation protocols, optimizing cell dosages, and implementing immunosuppressive regimens when necessary. Additionally, long-term monitoring and evaluation of patients undergoing stem cell-based therapies are essential to assess potential side effects, including tumorigenicity and immune responses (31, 66). From this perspective, the development of therapeutic cells has also involved “fail-safe approach” efforts in methods for controlling their fate. These endeavors include recognizing markers associated with cancerous transformations in cells that fail to complete their final differentiation or markers signifying a continuing proliferative state in cells remaining in a pro-mitotic state (67, 68). In such cases, cell engineering is employed to activate artificial cell suicide programs, facilitating self-destruction. Alternatively, more assertive techniques, such as gamma-ray irradiation, are being explored to induce damage specific to undifferentiated cells that still retain proliferative potential post-transplantation (69). These methods are currently under investigation for their potential utility as safety measures.
Immunological considerations: Another challenge in stem cell-based therapies is the potential for immune rejection of transplanted cells. ESCs, iPSCs, and NSCs may elicit immune responses in recipients, which can compromise the survival and functionality of transplanted cells. Strategies to address immunological compatibility include the use of immunosuppressive medications or the application of immune-matched or patient-specific stem cells. For example, successful transplantation of autologous hiPSC-derived mDA neurons has been reported, circumventing the need for immunosuppression (70). Further research, such as the development of hypo-immune hiPSCs (universal hiPSCs) (71), is necessary to optimize immunosuppression regimens and develop immunomodulatory approaches to enhance the long-term survival of transplanted cells.
Limited availability and scalability: The ability to apply stem cell-based therapies for PD on a broader scale is constrained by the limited availability and scalability of suitable cells (72). The challenge of producing high-quality and standardized cell populations for transplantation remains prominent. There is a need for scalable protocols to enable the large-scale production of mDA neurons or NSCs to meet the demands of potential clinical applications. Furthermore, establishing robust manufacturing processes, implementing quality control measures, and ensuring compliance with regulatory standards are essential steps for translating stem cell-based therapies into clinical practice.
Ethical and regulatory considerations: Stem cell-based therapies bring forth ethical and regulatory considerations, particularly when utilizing hESCs or fetal-derived cells (73). The use of hESCs raises ethical concerns related to the destruction of embryos (74). However, the development of hiPSCs provides an alternative source of patient-specific cells without ethical controversies. It is of paramount importance to strictly adhere to ethical guidelines and regulatory frameworks to ensure patient safety and the responsible advancement of stem cell-based therapies (75, 76).
Go to :
Stem Cell-Based PD Models and Drug Development
Harnessing stem cell technologies and employing cutting-edge approaches for disease modeling has led to the elucidation of pathophysiologies in various diseases (77). Moreover, it has resulted in the validation of molecular and cellular therapeutic targets (78, 79) and has advanced methodologies for drug discovery (80, 81), ultimately enhancing strategies for disease treatment (82).
Generation and utilization of stem cell-based PD models
Stem cell-based disease models have brought about a revolution in PD research. These models provide a platform to study the underlying mechanisms of the disease and facilitate drug development, as illustrated in Fig. 2.
Disease modeling studies utilizing hESCs have offered insights into disease mechanisms and potential therapeutic targets for PD. For example, Cooper et al. (83) successfully differentiated hESCs into mDA neurons, identifying dysregulation of the WNT signaling pathway as a potential contributing factor in PD pathology. Byers et al. (84) used hESC-derived mDA neurons to demonstrate the involvement of the unfolded protein response and endoplasmic reticulum (ER) stress in PD pathogenesis. These studies showcase the utility of hESC-based models in investigating disease mechanisms and identifying novel therapeutic targets. In recent years, there has been a notable upswing in the utilization of hiPSCs for the purpose of modeling PD. A significant emphasis within the realm of hiPSC models lies in the exploration of α-syn (SNCA) mutations and gene multiplications associated with PD (4, 84-86). Especially, hiPSC-derived mDA neurons from individuals afflicted with SNCA mutations revealed elevated levels of α-syn and the resulting deleterious consequences, such as oxidative stress and ER stress (84, 86-88). Furthermore, α-syn oligomers have been identified as disruptors of axonal transport, mitochondrial function, and synaptic connectivity in hiPSC-derived DA neurons (89, 90), as detailed in Table 1 (84, 87-117).
Table 1
Phenotypes of PD hiPSC-derived mDA neurons with genetic mutations
| Gene | Mutation | Phenotype in DA neurons | Reference |
|---|---|---|---|
| SNCA | 3X SNCA | Increased α-synuclein, oxidative stress, ER stress, disrupted protein response, impaired mitochondrial transport, neuronal death | (84, 87-92) |
| 2X SNCA | Elevated α-synuclein levels, aggregation and phosphorylation, impaired mitochondrial transport, increased oxidative stress | (89, 93) | |
| A53T | Altered mitochondrial morphology and function, defective neurite growth, axonal abnormalities | (91,92, 94-97) | |
| GBA1 | N370S | Decreased GBA activity, increased α-synuclein, lower dopamine synthesis, increased MAO-B expression, ER stress, autophagic and lysosomal disturbance | (98-100) |
| L444P, RecNcil | Dysfunctional NAD+ metabolism, altered mitochondrial function | (101) | |
| PARK2/PARKIN | Various (in early-onset PD) | Increased oxidative stress, ROS, abnormal neurotransmitter homeostasis, impaired mitophagy, increased apoptosis, mitochondrial dysfunction | (102, 103) |
| V324A | Accumulation of α-synuclein, abnormal mitochondria, increased vulnerability to mitochondrial toxin | (104) | |
| PINK1 | C568A | Impaired mitophagy | (105) |
| Q456X, V170G | Accumulation of α-synuclein, increased mitochondrial copy number, impaired ability to recruit PARKIN to mitochondria, upregulation of PGC-1α | (104, 106) | |
| LRRK2 | G2019S | Accumulation of α-synuclein, low mitochondrial respiration, increased oxidative stress, impaired mitophagy, dysregulated endosomal trafficking, altered autophagy, increased LRRK2 kinase activity | (107-116) |
| VPS35 | D620N | Decreased autophagic flux and lysosomal mass, accumulation of α-synuclein, apoptotic cell death, impaired mitochondrial function and respiration | (117) |
![]()
LRRK2 mutations, considered another genetic risk factor for PD, have undergone extensive examination via the use of hiPSC models (107, 108, 118, 119). These models have brought to light a spectrum of pathological characteristics linked to LRRK2 mutations, encompassing mitochondrial dysfunction, oxidative stress, and compromised autophagy (83, 109, 110, 120). Importantly, hiPSC-derived DA neurons have served as a valuable tool for screening potential therapeutic compounds in the context of LRRK2-related PD (121). Exploration of mutations in the GBA gene, known to heighten the risk of PD, has been undertaken employing hiPSC models (98, 99, 101). These models have furnished valuable insights into the role of GBA mutations in α-syn accumulation, lysosomal dysfunction, and mitochondrial impairment (100, 101). Furthermore, hiPSC-based investigations have uncovered promising therapeutic strategies for GBA-associated PD (122). Additionally, hiPSC models have been employed to validate other PD-related genes, including PINK1 (123), PARK2/PARKIN (104), and VPS35 (124). These investigations have unveiled critical facets of mitochondrial dysfunction, impaired mitophagy, and disrupted endo-lysosomal trafficking in the pathogenesis of PD (104, 117, 125). Beyond the study of monogenic forms of the disease, hiPSC models have significantly contributed to our comprehension of sporadic PD (126-128). They have illuminated aberrations in oxidative stress, epigenetic modifications, and proteostasis pathways, offering valuable insights into the intricate nature of sporadic PD. Collectively, these models hold tremendous promise for advancing our understanding of PD pathogenesis, the identification of novel therapeutic candidates, and the rigorous evaluation of their effectiveness, constituting a pivotal component of PD research.
Advantages and limitations of stem cell technology in drug development
Stem cell technology presents several advantages in the context of PD drug development. One of the key benefits is the ability to generate disease-relevant cell types affected in PD. For instance, hiPSCs derived from PD patients can be differentiated into mDA neurons, enabling the investigation of disease-specific mechanisms. Utilizing this approach, numerous groups have revealed PD-associated phenotypes in mDA neurons and identified therapeutic compounds capable of alleviating the phenotypes, as detailed in Table 2 (83, 95, 99, 121, 122, 125, 127-139).
Table 2
Overview of PD hiPSC-based models utilized for drug discovery
| Gene | Phenotype in DA neurons | Treatment | Effect of treatment | Reference |
|---|---|---|---|---|
| SNCA | Increased intraneuronal nitric oxide and protein nitration, mitochondrial dysfunction | L-NAME (an inhibitor of nitric oxide synthase), isoxazole (activates MEFC2) | Decreased nitric oxide, increased MEFC2 activity | (95) |
| SNCA | Defective neurite outgrowth, fragmented axons with swollen structures, disrupted synaptic connectivity | NPT100-18A, NPT100-14A, ELN48228 | Rescued phenotypes | (129) |
| SNCA | Increased levels of α-synuclein | Stearoyl CoA desaturase | Reduced α-synuclein levels | (130) |
| SNCA | Increased α-synuclein levels | A-443654 (AKT modulator) | Normalized α-synuclein levels | (131) |
| SNCA | Increased α-synuclein levels | ML348 (lysophospholipase 1 inhibitor) | Reduced total and phosphorylated α-synuclein levels | (132) |
| SNCA | Increased intracellular α-synuclein levels, reduced D2 receptors availability, dysregulated firing activity | Quinpirole (a D2 and D3 receptor agonist) | Restored defective firing activity | (133) |
| SNCA, GBA, PARK9 | Presence of amyloidogenic α-synuclein, the accumulation of α-synuclein, and reduced neuronal viability | NCGC758 | Improved GCase activity and reduced α-synuclein levels | (134) |
| GBA | Decrease in GBA activity, increased levels of α-synuclein | NCGC607 | Stabilized the unstable mutant enzyme, restored GBA activity and protein levels, reduced glycolipid storage, decreased α-synuclein levels | (134) |
| GBA | GBA enzymatic activity of ~50%, increased level of α-synuclein, lower capacity to synthesize and release dopamine, increased MAO-B expression | MAO-B inhibitors | Normalized both α-synuclein and dopamine levels | (99) |
| GBA-84GG, LRRK2, DJ-1, PARKIN | ER stress, autophagic and lysosomal disturbance, presence of extracellular α-synuclein | S-181 | Increased WT GBA activity, restored lysosomal function, lowered accumulation of glucosylceramide, oxidized dopamine, and α-synuclein | (122) |
| GBA, LRRK2 | Defective NAD+ metabolism, altered mitochondrial functions, lower GBA levels, increased α-synuclein levels, accumulation of oxidized dopamine | Nicotinamide riboside, quetiapine | Rescued mitochondrial functions, partially rescued GBA levels, reduced α-synuclein levels and accumulation of oxidized dopamine | (135) |
| PARKIN | Ubiquitin system deficiencies in microtubules | Taxol |
Normalized neuronal morphology Microtubule stabilization |
(136) |
| PARKIN | Rotenon-induced oxidative stress, apoptosis, mitochondrial dysfunction | Benidipine | Rescued rotenon-induced stress | (121) |
| PARKIN, PINK1 | Mitophagy abnormalities | Flunarizine | Rescued mitochondrial functions | (137) |
| PARKIN |
Mitochondrial dysfunction Ubiquitin system deficiencies |
NC001-8 (indole derivative) | Increased NRF2 antioxidative pathway | (137) |
| PARKIN, PINK1 | Defective PINK1/PARKIN-mediated mitophagy | Memantine | Increased clearance of damaged mitochondria | (125) |
| LRRK2, PINK1 | Increased susceptibility to oxidative stress, mitochondrial dysfunction, increased apoptosis | Coenzyme Q10, rapamycin, GW5074 (LRRK2 kinase inhibitor) | Reduced cell vulnerability to oxidative stress | (83) |
| LRRK2 | Increased vulnerability of DA neurons to immune challenge, shortening of DA neurites | Interferon-γ | Enhanced LRRK2-G2019S-dependent negative regulation of AKT phosphorylation and NFAT activation | (138) |
| Young onset PD | Increased accumulation of α-synuclein and phosphorylated protein kinase Cα, reduced abundance of lysosomal membrane proteins | PEP005 | Reduced α-synuclein and phosphorylated protein kinase Cα levels while increasing LAMP1 abundance | (127) |
| Idiopathic PD, DJ-1 KO | Accumulated neuromelanin and oxidized dopamine, lysosomal dysfunction | mito-TEMPO, NAC | Reduced the accumulation of oxidized dopamine and improved lysosomal GCase activity and proteolysis | (128) |
| PLA2G6 D331Y | ER stress, abnormal calcium homeostasis, mitochondrial dysfunction, increased reactive oxygen species, and apoptosis via restoring the ER function and CREB signaling | Azoramide | Rescued phenotypes | (139) |
![]()
Additionally, stem cell-based models facilitate high-throughput screening assays and validation processes for identifying potential therapeutic compounds. For example, Tabata et al. (121) conducted a high-throughput screen on hiPSC-derived mDA neurons and discovered compounds that suppressed rotenone-induced apoptosis while promoting neuronal survival, suggesting their potential as therapeutic agents. Similarly, Yamaguchi et al. (137) utilized a semi-automated phenotypic analysis system to screen a library of compounds using hiPSC-derived mDA neurons from familial PD patients. They successfully identified four compounds that improved mitochondrial clearance and enhanced the functionality of mDA neurons.
However, despite its advantages, stem cell technology encounters certain limitations that require addressing for its effective application in drug discovery. One such limitation pertains to the complexity of modeling a multifactorial disease like PD, which entails intricate interactions between various cell types and environmental factors (140). To address this challenge, recent studies have concentrated on the development of multicellular systems, such as three-dimensional (3D) organoid models created from hiPSCs. For example, Kwak et al. (141) engineered a 3D midbrain organoid model, enabling exploration into the effects of midbrain-related cell types on the survival of mDA neurons and providing valuable insights into disease mechanisms. Another obstacle lies in maturing stem cell-derived mDA neurons to faithfully replicate the PD phenotypes observed in neurons resembling those in PD patients. To tackle this issue, diverse strategies have been explored, including extended culture periods (142), co-cultivation with supporting cells (143, 144), and genetic manipulation (145). Furthermore, the variability observed among hiPSC-derived neuronal cultures and differentiation protocols complicates the generation of consistent and reproducible results, posing challenges for the development of high-throughput screening assays. Efforts have been made to standardize protocols and optimize differentiation conditions to reduce variability and enhance the reliability of stem cell-based models. For instance, Nolbrant et al. (146) devised a defined and scalable differentiation protocol for generating mDA neurons from hiPSCs, which demonstrated improved efficiency and consistency across different hiPSC lines.
The need for a novel stem-cell based PD pathogenesis model or drug discovery platform
The enactment of the U.S. Food and Drug Administration (FDA) Modernization Act 2.0 in December 2022 has underscored the imperative need for non-animal testing alternatives in drug development (147, 148). This regulatory shift promotes the utilization of human-centric models, such as hiPSC technology and organoids, for non-clinical tests in the field of drug development and safety evaluations. Consequently, there exists an urgency to devise advanced hiPSC-based models capable of accurately recapitulating human disease pathology and offering enhanced preclinical assessment of drug candidates, aligning with the FDA’s strategic objectives. While PD is a multifaceted disorder with various contributory factors, including genetic mutations, environmental influences, oxidative stress, and mitochondrial dysfunction, a common hallmark of PD is the accumulation of α-syn proteins within mDA neurons. This molecular event remains a fundamental feature of PD pathogenesis, leading to the formation of α-syn aggregates known as Lewy bodies. These aggregates are believed to selectively exert toxicity in mDA neurons, resulting in neuronal dysfunction and eventual death-key characteristics of PD (1, 2, 5). Given the central role of α-syn aggregation in PD, there is a pressing necessity for a novel hiPSC-based model to expedite the development of interventions that can impede or reverse the progression of this debilitating disorder. However, substantial challenges persist, primarily due to the somatic reprogramming involved in generating hiPSCs, which resets the cells to an embryonic state and makes it challenging to model late-onset diseases like PD characterized by α-syn aggregation (149, 150). Consequently, additional strategies are required to accurately replicate the aberrant protein aggregation intrinsic to the disease progression (151, 152). Furthermore, it is crucial to recognize that most non-cell-based compound screenings suffer from limited reproducibility in human neurons, thereby underscoring the urgent need to establish reliable, consistent, and cost-effective human neuronal models for high-throughput drug discovery platforms. In conclusion, the establishment of a stem cell-based model focusing on α-syn aggregates in PD is of utmost importance. Such a model promises not only to enrich our comprehension of the disease but also to expedite the discovery of potential treatments, marking a significant advancement in the battle against PD.
Advantages, limitations, future integration, and direction of the OASIS model
To address these challenges, we have recently developed the OASIS model (153, 154). This model presents several advantages for studying PD and sheds light on its potential integration with other technologies and future directions (Fig. 3). One key advantages of the OASIS model is its unique ability to induce α-syn aggregation using optogenetic proteins within a short timeframe. This distinctive feature allows precise control and manipulation of the formation of pathological α-syn aggregates, closely resembling the Lewy bodies observed in PD. By investigating the impact of α-syn aggregation on cellular processes and neurodegeneration, the OASIS model offers valuable insights into the underlying mechanisms of PD pathogenesis. Another advantage of the OASIS model is its provision of a simplified, cost-effective platform for drug screening and the identifying potential therapeutic candidates. Through controlled induction of α-syn aggregation, compounds can be efficiently screened to reduce aggregate formation, yieling highly consistent results. Moreover, the OASIS model employs hiPSC-derived mDA neurons and midbrain organoids from PD patients, enabling the generation of patient-specific multiple cell types. This allows for a more accurate assessment of the effectiveness of potential hit compounds in reducing cellular toxicity and restoring normal function. As such, the OASIS model serves as a non-clinical testing platform for PD, holding promise for discovering new therapeutic targets and developing disease-modifying treatments.
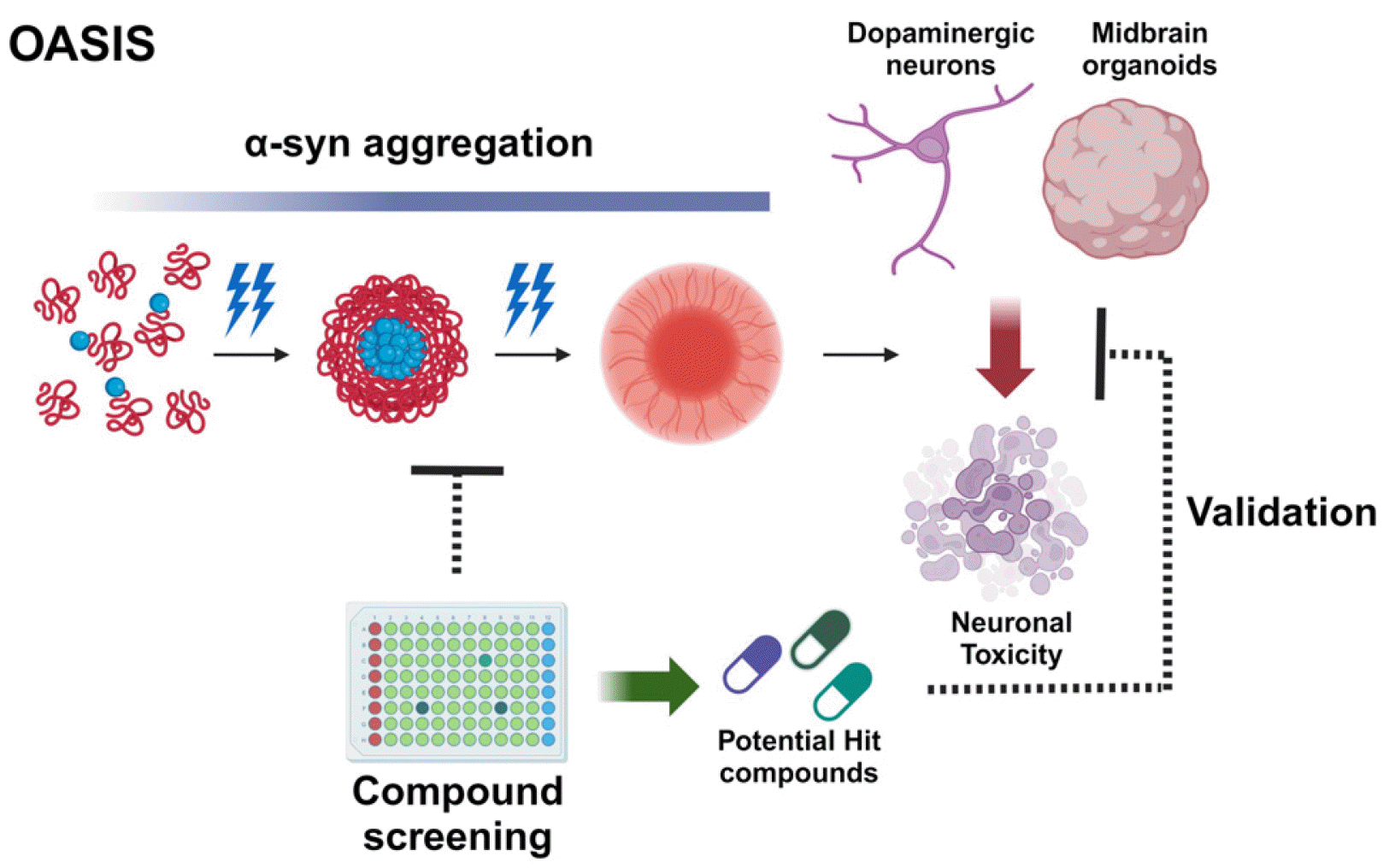
Nevertheless, it is crucial to consider the limitations of the OASIS model. One constraint is its primary focus on the aggregation of α-syn in SNCA triplication PD iPSCs. Another concern is its exclusive emphasis on α-syn aggregation, despite PD involving a complex interplay of diverse cellular processes originating from other genetic mutations or sporadic PD patients’ hiPSCs. These aspects are linked to the limitations of the OASIS model, which, despite its ability to mimic α-syn aggregation, is confined to representing the molecular phenotype at the endpoint of cellular pathology. The model faces limitations in depicting the cellular pathological mechanisms initiating PD, including the recently investigated propagation of misfolded α-syn in the gut-brain axis and functional impairment due to misfolded α-syn within cells (155, 156). Falling short, the model fails to capture the broader molecular pathological mechanisms occurring before α-syn aggregation, such as issues stemming from a low functional protein dose of α-syn (157) and dominant-negative mutants (158) caused by α-syn mutations (159, 160). This underscores the necessity for model improvements to achieve a more precise understanding of molecular and cellular pathology related to the α-syn function on various biological events (161-164), as well as a more comprehensive understanding that encompasses the spread of pathological molecular features in PD.
Utilizing the OASIS model based on patient-specific hiPSCs facilitates the exploration of pathogenic mechanisms reflecting the individual genetic profiles, encompassing various genetic mutations implicated in PD onset. This approach allows for the identification of personalized therapeutic interventions and assessment of their potential efficacy, making it possible to evaluate individual responses to treatment and optimize therapeutic strategies. Conversely, introducing diverse SCNA mutations into normal cells with the same genetic background and utilizing OASIS advances knowledge regarding the penetrance and diversity of PD onset and symptoms for each genetic mutation type, contributing to a precise understanding of PD pathogenesis. Moreover, the OASIS model can be enhanced by integrating with other advanced technologies, such as microfluidic systems (165), organoids (166, 167), or organ-on-a-chip platforms (168), enabling the mimicry of the physiological environment of PD pathology-relevant tissues. This allows for the exploration of cell-cell interactions, neuronal circuitry, and synaptic activity within the context of α-syn aggregation. Additionally, incorporation of advanced imaging techniques and multi-omics approaches can provide a more detailed characterization of cellular and molecular changes associated with PD.
In conclusion, the OASIS model offers advantages such as precise control over α-syn aggregation, utilization of patient-specific hiPSCs, and potential for drug screening. However, it is crucial to carefully address the limitations of this model and consider future integration with other technologies to enhance its validity and comprehensiveness. The direction of the OASIS model lies in the potential development of personalized medicine and targeted therapies for PD. Depending on the extent of improvements, it can also contribute to the development of various pathological models and the elucidation of fundamental mechanism of PD onset and progression.
Go to :
Conclusion Remarks
Stem cell-based approaches offer immense potential for advancing the treatment of PD. The significance of stem cell technology in PD treatment lies in its capacity to address the causative path of the disease, provide relevant models for drug discovery, and enable personalized therapeutic strategies.
Stem cell-based strategies have the potential to revolutionize PD treatment in three significant ways. First, by replacing damaged mDA neurons, stem cell transplants can directly combat PD’s neurodegeneration, yielding promising early results. Second, these technologies can generate disease-specific models using patient-derived hiPSCs, accelerating the drug discovery process by furnishing accurate disease representations for testing potential therapies. Lastly, stem cells open the door to personalized medicine possibilities by generating patient-specific disease models, facilitating individualized treatment strategies. Despite existing challenges, such as enhancing the safety of stem cell transplants, the ongoing progress in stem cell technology provides hope for innovative treatments that can enhance the quality of life for PD patients.
Go to :
 XML Download
XML Download