

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Urothelial cancer (UC) develops from the epithelial cells that line the urinary tract.1 The vast majority of UC cases, over 90%, are found in the bladder, while the remaining cases originate in the ureter, urethra, or renal pelvis.2 Bladder cancer (BC) is the 10th most common malignancy globally in 2020, with an annual incidence of approximately 573,000 new patients and 212,536 deaths.3 According to the Korea National Cancer Incidence Database (KNCI DB) and the International Agency for Research on Cancer (IARC), the age-standardized incidence rates of BC per 100,000 population in Korea in 2020 were 7.5 and 7.9 for men and 1.4 and 1.5 for women, respectively. In comparison, global incidence rates reported by the IARC were 9.5 for men and 2.4 for women.45 In Korea, the annual number of BC cases nearly tripled between 2000 and 2020, rising from 1,744 to 4,753 cases (KNCI DB data). The incidence rate was five times higher in men than in women. Additionally, the number of BC-related deaths in Korea doubled during the same period, increasing from 778 to 1,593 deaths per year.6 The 5-year survival rate for BC patients diagnosed in Korea between 2016 and 2020 was 76.5%.4
BC can be divided into three distinct categories, each with unique prognosis, management strategies, and treatment objectives. The first category comprises patients with non-muscle-invasive bladder cancer (NMIBC), in which the focus of treatment is to minimize recurrence and prevent advancement to more severe stages. The second category includes patients with muscle-invasive bladder cancers (MIBCs). Unlike NMIBC, MIBC has a higher risk of progression and requires more intensive treatment, typically involving a multidisciplinary strategy that combines systemic treatment, surgical interventions, and/or radiation therapy. The final category involves metastatic bladder cancer (mBC), where the primary concern is to extend patient survival and enhance the quality of life. Various therapeutic agents with diverse mechanisms are employed to combat this form of disease and to improve both survival rates and quality of life.7
For over four decades, cisplatin has been the primary treatment for metastatic urothelial cancer (mUC), but only half of the patients are eligible for it, and the survival benefits are modest.8 Recently, immune checkpoint inhibitors (ICIs) such as atezolizumab and pembrolizumab have shown efficacy in mUC, offering significant clinical benefits.9 However, there is a continuous demand for new treatments. The Cancer Genome Atlas (TCGA) project identified various genomic alterations in UC, leading to the development of targeted therapies.10 Fibroblast growth factor receptors (FGFRs) play a crucial role in mUC, with FGFR2 and FGFR3 being particularly significant.11 FGFRs, which comprise four transmembrane receptors (FGFR1–4), are involved in cell proliferation, survival, and migration. Alterations in FGFRs such as gene amplification, mutations, and fusions have been observed in various cancers, including UC, with FGFR3 alterations being particularly prevalent in UC.12
The treatment landscape for mUC has changed over time. Based on the outstanding results of the EV-302 trial, the National Comprehensive Cancer Network recommends the combination of enfortumab vedotin (EV) with pembrolizumab as the primary systemic treatment for patients with locally advanced (la)/mUC, irrespective of their eligibility for cisplatin.1314 Another treatment strategy, erdafitinib, which targets FGFR, has emerged as a treatment option, particularly for patients with FGFR3 mutations or fusions in la/mUC.
This review highlights the role of FGFR genomic alterations in UC and discusses the therapeutic potential of FGFR inhibition, ongoing clinical trials, and future challenges in targeting fibroblast growth factor (FGF)/FGFR signaling pathway.
Go to : 
OVERVIEW OF FGF/FGFR SIGNALING
FGFs are a group of broad-spectrum mitogens that are pivotal in the regulation of a plethora of cellular functions, including cell migration, proliferation, differentiation, and survival. These factors are instrumental in the development, metabolism, and maintenance of tissue homeostasis.15 Dysfunctions within the FGF or FGFR signaling axis are linked to various human diseases, including congenital disorders such as craniosynostosis and dwarfism, chronic conditions such as kidney disease, obesity, insulin resistance, and diverse types of tumors (Fig. 1).16
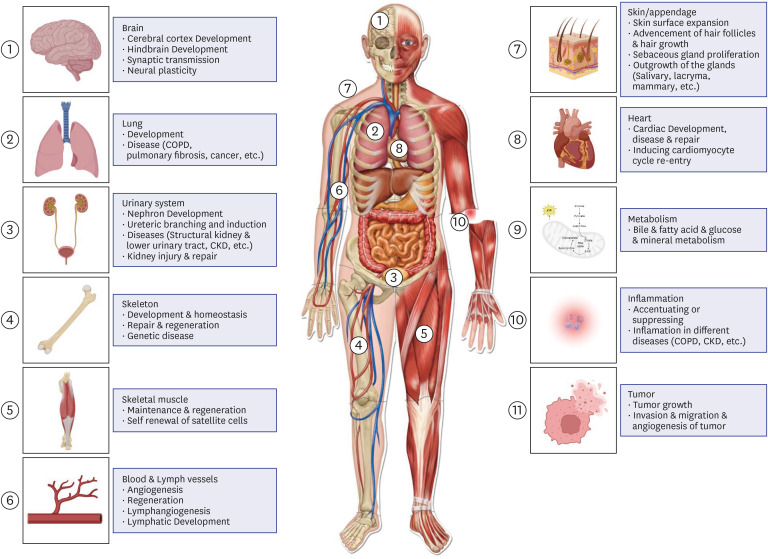 | Fig. 1Summary of the primary functions of FGF/FGFR signaling in various physiological and pathological contexts is as follows: FGF/FGFR signaling is crucial in the development of nearly all organs, including the lungs, heart, urinary system, brain, skeleton, muscles, and skin/appendages, along with angiogenesis and lymphangiogenesis. FGFs/FGFRs significantly influence tissue repair, regeneration, and inflammatory responses. Endocrine FGFs are vital in metabolism, affecting the kidneys, liver, brain, intestines, and adipose tissue. Disruptions in FGF/FGFR signaling can result in a variety of diseases, such as genetic disorders, cancer, COPD, and CKD. Created with BioRender.com.FGF = fibroblast growth factor, FGFR = fibroblast growth factor receptor, CKD = chronic kidney disease, COPD = chronic obstructive pulmonary disease.
|
The FGF family is one of the most diverse groups of vertebrate growth factors, and 22 FGF ligands have been identified in mice and humans. Based on sequence homology and phylogenetic analyses, these ligands have been categorized into six subfamilies: five paracrine subfamilies and one endocrine subfamily. The endocrine subfamily uniquely signals in a hormonal manner, whereas the paracrine subfamily is involved in local signaling.17
FGFs deliver multifaceted biological effects by attaching to and stimulating high-affinity receptors with tyrosine kinase activity, which are encoded by four genes (FGFR1 to FGFR4), along with FGFRL1, which is a modified version of the receptor lacking an intracellular segment. These receptors are characterized by their single-pass configuration through the cell membrane, featuring domains external to the cell, spanning the membrane, and inside the cell that facilitate tyrosine kinase activity. Specifically, the outer segment includes a trio of immunoglobulin-like domains, an area of acidic amino acids, and a region that binds heparin, which is necessary for FGFs, along with heparan sulfate and various interacting proteins. The transmembrane domain is crucial for securing the receptors within the cellular membrane and enabling them to be paired. Within the cell, the region adjacent to the membrane aids in pairing receptors, whereas the split kinase domains are vital for propagating signals related to FGF.12
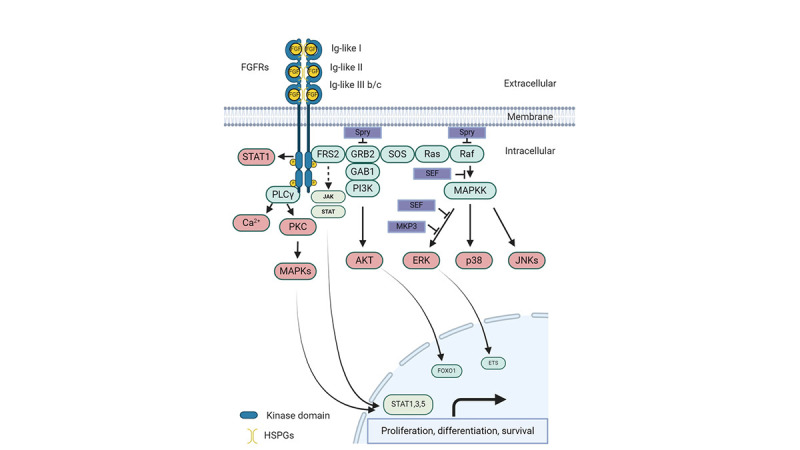
When FGFs bind to inactive FGFR monomers, this interaction prompts a series of structural changes in FGFRs, leading to their dimerization and subsequent activation of intracellular tyrosine kinases through the phosphorylation of tyrosine residues in the cytosolic tails of FGFRs.18 Following this, the phosphorylated tyrosine sites become attachment points for other signaling proteins, including FGFR substrate 2α, situated on the cell membrane. Additionally, FGFRs engage and phosphorylate the SH2 domain-inclusive phospholipase Cγ (PLCγ), forming a 2:1 FGFR-PLCγ allosteric complex, demonstrating the essential role of FGFR dimerization in the phosphorylation of substrates. The typical downstream pathway of FGF/FGFR signaling in different cells and tissues encompasses the Ras/Raf-MEK-MAPK (mitogen-activated protein kinase) cascade, the PI3K/AKT pathway, PLCγ, and the signal transducer and activator of transcription. Moreover, several proteins linked to the FGF syn-expression group have been identified, including sprouty, XFLRT3, SEF, and MKP3. These proteins are regulated by FGF signaling and are closely co-expressed with FGFs, often acting to restrain FGF/FGFR signaling via negative feedback mechanisms (Fig. 2).16
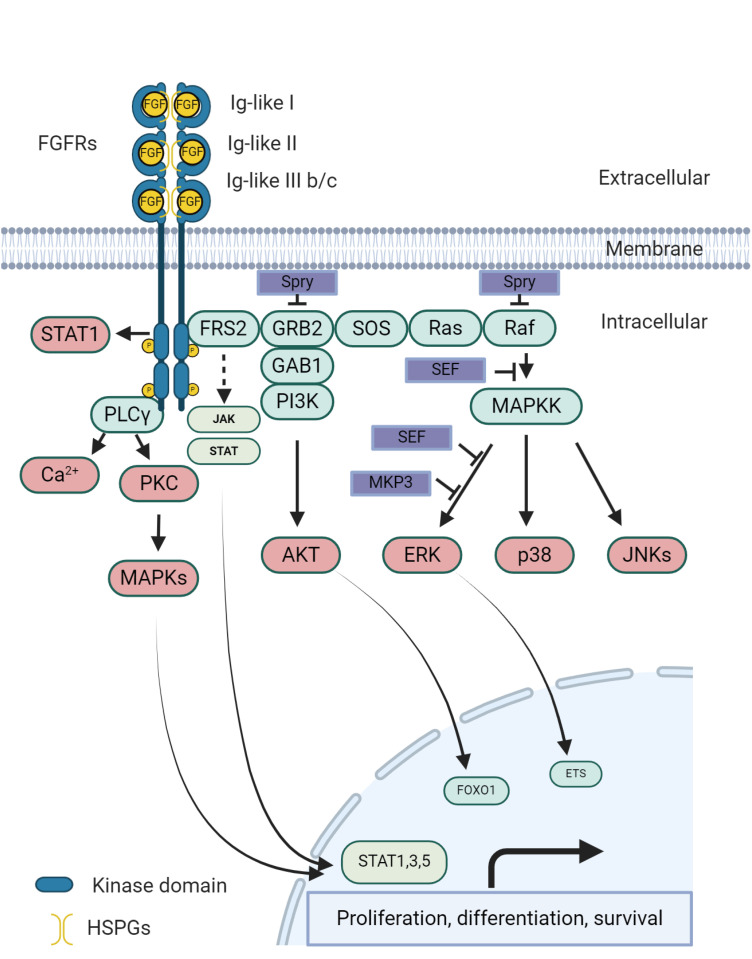 | Fig. 2The classical FGF/FGFR pathways involve several key steps and molecules. When appropriate growth factors bind to receptors, they induce conformational changes in FGFRs, leading to their dimerization and activation. Once activated, FGFRs phosphorylate FRS2α, which then binds to the SH2 domain-containing adaptor protein Grb2. Grb2 connects to SOS, GAB1, and Cbl through its SH3 domain, initiating the activation of the Ras/Raf/MAPK pathways, including ERK MAPK, p38 MAPK, and JNK MAPK. Additionally, activated FGFRs stimulate phosphatidylinositol-3 kinase and STAT pathways. FGFRs also recruit and phosphorylate PLCγ. Within the FGF synexpression group, SEF and XFLRT3, both transmembrane proteins, directly interact with FGFRs. SEF acts as a negative regulator by modulating the phosphorylation of the MAPK ERK cascade, while XFLRT3 enhances FGF/FGFR signaling by forming a complex with FGF receptors. Spry modulates FGF/FGFR signaling by acting at the level of Grb2 and/or Raf, and MKP3 negatively regulates this signaling by dephosphorylating activated ERK. Created with BioRender.com.FGF = fibroblast growth factor, FGFR = fibroblast growth factor receptor, MAPK = mitogen-activated protein kinase, PLCγ = phospholipase Cγ, FRS2α = fibroblast growth factor receptor substrate 2α, GAB1 = growth factor receptor-bound 2-associated binding protein 1, GRB2 = growth factor receptor-bound 2, PKC = protein kinase C, SOS = son of sevenless.
|
The variety of roles played by FGF/FGFR signaling underscores its complex regulatory mechanisms. Modulation of FGF/FGFR signaling occurs at multiple stages, including the specificity of ligand-receptor interactions, gene expression, and alternative splicing. Additionally, interactions between FGF/FGFR and other signaling cascades, such as bone morphogenetic protein and Wnt, modify signaling processes. The specific or varied binding capacities of FGF/FGFR, which are influenced by ligand-driven receptor alignment, are crucial for finely tuning FGF-mediated signaling. Spatial and temporal variations in the expression of FGFs, FGFRs, and heparan sulfate cofactors play critical roles in the stringent control of this signaling pathway.19 The diverse distribution of these signaling components across different tissues and their varying expression levels significantly affect tissue development, maintenance, and disease progression. The creation of multiple FGF/FGFR isoforms through alternative splicing and translational initiation, such as the specific splicing at domain D3 of FGFR1, FGFR2, and FGFR3 that results in b and c isoforms, influences their functional specificity across various cells and tissues. Moreover, the expression and signal characteristics of FGF/FGFR components are further regulated by epigenetic factors and post-translational modifications, such as phosphorylation, glycosylation, ubiquitination, and the cellular movement of these molecules.16
Remarkable progress has been made in recent decades in understanding the modulation of the FGF/FGFR signaling cascades. Such advancements deepen our understanding of this complex signaling network and provide opportunities for novel therapeutic interventions targeting aberrant FGF/FGFR signaling pathways. With FGFR5, the newest member lacking an intracellular domain, the potential for new receptor interactions and signaling outcomes is a subject of ongoing research, highlighting the dynamic nature of this signaling axis.20
Go to :
FGFR PATHWAY IN UC
Genomic alterations in FGFRs have been extensively studied in various human cancers.19 Helsten et al.21 reported that among approximately 4,000 tumors analyzed, 7.1% exhibited alterations in FGFRs. In UC, FGFR3 somatic mutations occur in 15% of patients, FGFR1 amplification in 7%, and gene fusions in 6%. The American Association for Cancer Research Project Genomics Evidence Neoplasia Information Exchange database v13.0-public further illustrates the frequency of FGFR1–4 alterations in UC, including both known drivers and alterations of unknown significance.22
A study by Kamoun and colleagues23 on the molecular classification of UC highlights the landscape of UC subtypes, detailing frequent mutations, oncogenic alterations, histology, and associated clinical phenotypes. FGFR3 dysregulation, through mutation, overexpression, or both, was observed in 54% of invasive UCs. FGFR3 mutations are particularly prevalent (~80%) in noninvasive UCs and upper tract UCs, which are often luminal papillary gene expression subtype tumors with poor immune infiltration. FGFR3 mutations are implicated in 5–20% of patients with MIBC. These mutations typically occur in the extracellular region and lead to ligand-independent dimerization, activation, and signaling. Invasive tumors are more likely to exhibit upregulation of wild-type FGFR3, which may lead to ligand-independent dimerization and activation.23
FGFR3-TACC gene fusions relevant to MIBC are more common in younger patients, never-smokers, and those of Asian ethnicity. These fusions, formed via tandem duplications on chromosome 4p16, are relatively rare (2–3% in MIBC) but significant. These mutations alter TACC3 function, potentially causing mitotic defects and aneuploidy.24
FGFR1 genomic alterations, although less studied than FGFR3 alterations, have a prevalence of 7–14% in various cancer types.21 FGFR1 and subsequent MAPK activation promote proliferation, survival, and epithelial-to-mesenchymal transition.25 Resistance to FGFR inhibition can develop through the upregulation of bypass pathways or gatekeeper mutations in the FGFR-binding domain.26 Specific FGFR inhibitors, such as Debio 1347 and futibatinib, are effective against these resistant mutations.27
TCGA and the Bladder Cancer Taxonomy Group have classified UC into several subtypes based on gene expression analyses. TCGA has identified four subtypes: luminal cluster I, luminal cluster II, basal cluster III, and basal cluster IV, with luminal cluster I commonly harboring FGFR3 mutations.28 The Bladder Cancer Taxonomy Group has identified six molecular classes of MIBC, with luminal papillary tumors frequently exhibiting FGFR3 translocations and mutations.23 This classification is crucial for understanding potential therapeutic options through the inhibition of the FGFR pathway in patients with la/mUC.
The FGFR signaling pathway, which involves transmembrane proteins with intracellular tyrosine kinase domains derived from the FGFR1–4 genes, plays critical roles in cell growth, angiogenesis, and proliferation. Alterations and fusions of FGFR3 activate downstream pathways such as MAPK and PI3K/AKT, contributing to cancer development and progression.29 This understanding has led to the development of targeted inhibitors, such as erdafitinib.
FGFR mutations in UC exhibit distinct patterns depending on the disease stage, ranging from organ-confined to metastatic states. In early stages, particularly in NMIBC, FGFR3 mutations are highly prevalent and are often associated with low-grade tumors and favorable prognoses.30 These mutations suggest that FGFR3 plays a significant role in the early development of UC, contributing to a more indolent form of tumorigenesis. As the disease progresses to more advanced stages, such as muscle-invasive or locally advanced UC, the frequency of FGFR mutations, particularly FGFR3, decreases. This shift is accompanied by the emergence of other genetic alterations that drive the tumor’s aggressiveness and metastatic potential.31 In mUC, FGFR mutations are less common but remain a critical target for therapy, especially in cases where other oncogenic drivers are absent. FGFR inhibitors, particularly in patients with FGFR2 or FGFR3 alterations, have shown efficacy in prolonging progression-free survival (PFS), highlighting their therapeutic potential even in advanced stages.32 However, the effectiveness of FGFR3 alterations as predictive markers for response to platinum-based chemotherapy is less clear. Studies indicate that FGFR3 mutations do not significantly influence overall survival (OS) or PFS in patients treated with first-line platinum-based chemotherapy, suggesting that these alterations are neither predictive nor prognostic for chemotherapy response.193334353637 Nonetheless, FGFR3 mutations remain important for identifying patients who might benefit from targeted therapies after chemotherapy has failed, underscoring the complex and evolving role of FGFR mutations in the clinical management of UC.1938
Go to :
CLINICAL DEVELOPMENT OF FGFR INHIBITORS IN UC
First-generation FGFR inhibitors
First-generation FGFR inhibitors are nonselective tyrosine kinase inhibitors (TKIs) that compete with ATP for ATP-binding sites.29 This competition inhibits the function of FGFRs, which are crucial in cellular processes like proliferation and survival.39 The development of these inhibitors has targeted cancers with FGFR aberrations. However, the development of resistance to these inhibitors has led to the development of next-generation covalent inhibitors to overcome this issue. Despite the complexity of their mechanisms, most FGFR inhibitors function by binding to the adenine binding site of the receptor tyrosine kinase (RTK).29 First-generation and promiscuous FGFR TKIs, such as dovitinib, nintedanib, and lenvatinib, have been tested alone or in combination with cisplatin-based chemotherapy or PD1 inhibition in unselected patients. Although nintedanib did not enhance the pathologic complete response in combination with neoadjuvant cisplatin, it improved PFS and OS. Lenvatinib, which also inhibits vascular endothelial growth factor receptors (VEGFRs), showed promise in combination with pembrolizumab as a first-line therapy, leading to a phase III trial to compare pembrolizumab with lenvatinib plus pembrolizumab in patients ineligible for platinum therapy with programmed death-ligand 1 (PD-L1) high-expressing tumors.404142
Erdafitinib
Pharmacokinetics
Erdafitinib, an orally administered pan-FGFR TKI targeting FGFR1–4, effectively inhibits the FGFR signaling pathway through its accumulation in intracellular lysosomes.43 This mechanism is supported by pre-clinical studies showing the antitumor effects of erdafitinib in xenograft models derived from human tumor cells and tissues of patients with cancer.44 Pharmacokinetic analysis of erdafitinib demonstrated a direct correlation between the administered dose and both the peak plasma concentration and the area under the curve for serum concentration over time.45 On average, the effective half-life of the drug ranges from 42 to 72 hours, with a steady state achieved within 14 days.46 The drug undergoes metabolism primarily through the CYP2C9 and CYP3A4 enzymes and is excreted predominantly in feces (69%) and, to a lesser extent, in urine (19%).47 The average volume of distribution was small (28.8 L), with 99.8% of erdafitinib binding to plasma proteins, especially α1-acid glycoprotein. Early-stage research indicated that erdafitinib exhibits a low half-maximal inhibitory concentration ranging from 0.1 to 129.2 nm in cancer cells with FGFR mutations, indicating a strong preference for targeting this specific signaling pathway.48
Erdafitinib in platinum-refractory UC
Based on these results and a phase I study of patients with advanced solid tumors harboring FGFR mutations, erdafitinib was further evaluated in a phase II BLC2001 study involving patients with la/mUC.49 This study included patients with specific FGFR translocations or mutations who progressed after chemotherapy, and a few patients had received prior immunotherapy. The study design allowed for dose adjustments based on tolerability, with the primary goal of assessing response rates and secondary outcomes such as PFS and OS, response duration, and biomarker-specific responses.
Approximately 24% of these patients had been treated with immunotherapy, while a smaller group (12%) did not receive chemotherapy because of contraindications to cisplatin. The selection criteria for participants included the presence of FGFR3 mutations (R248C, S249C, G370C, and Y373C) or FGFR2/3 gene fusions (FGFR3-TACC3, FGFR3-BAIAP2L1, FGFR2-BICC1, and FGFR2-CASP7) identified using the RT-PCR QIAGEN test on stored tumor samples. FGFR3 mutations were the most common genetic alterations observed in 69% of patients, followed by FGFR2/3 fusions in 25%, and a combination of mutations and fusions in 6%. Initially, 111 patients were randomized to receive erdafitinib under two dosing schedules: continuous (6 mg daily) or intermittent (10 mg for 7 days followed by a 7-day break). After an interim efficacy assessment, the study was adjusted to a single-arm format, where 101 patients were administered a continuous 8 mg daily dose, and the intermittent dosing group was discontinued.
Serum phosphate levels were monitored as a marker of FGFR3 inhibition, aiding in dosage personalization. Following phase I findings, if a patient’s serum phosphate was ≤ 5.5 mg/dL (1.8 mmol/L) by day 14, the dose of erdafitinib was increased to 9 mg daily, provided there were no significant treatment-related side effects. At 11 months and upon further evaluation at 24 months, the BLC2001 trial demonstrated an overall response rate (ORR) of 40%, including 36% partial and 4% complete responses. The average time to observe a response was 1.4 months, with a median PFS of 5.5 months and an OS of 11.3 months. Further analyses showed that the benefits of erdafitinib in terms of PFS and OS were consistent across subgroups, irrespective of the presence of visceral metastases or prior treatment with chemotherapy or immunotherapy. Despite its efficacy, two-thirds of participants experienced significant adverse events (AEs), including hyperphosphatemia, which may serve as a biomarker of treatment response. Given its significantly higher response rate compared to standard immunotherapy or second-line chemotherapy, the success of erdafitinib in the BLC2001 study underpinned its expedited approval by the U.S. Food and Drug Administration (FDA) on April 12, 2019, making it the sole FGFR inhibitor endorsed for mUC treatment.50
Erdafitinib versus chemotherapy in advanced or mUC
The THOR trial (BLC-3001/NCT03390504), a phase 3 confirmatory randomized study, explored the efficacy of erdafitinib versus the choice of chemotherapy determined by investigators in individuals with mUC who had worsened following one or two previous therapies, including treatment with an anti-PD-(L)1 drug.51 Participants in this study, all of whom had undergone one or two rounds of systemic therapy and had specific FGFR3 or FGFR2 alterations (mutations or fusions), were enrolled in the first cohort. Specifically, adults aged 18 years and older with inoperable la/mUC, carrying specific FGFR3 or FGFR2 alterations, an ECOG performance status of 0–2, satisfactory organ functionality, showing progression following prior systemic therapy, including anti-PD-(L)1 medication, and having received no more than two prior therapy lines, were randomized in a 1:1 ratio. Patients were treated with either erdafitinib (8 mg daily, with a potential increase to 9 mg on day 14, based on pharmacodynamics) or the investigator’s chemotherapy selection (docetaxel or vinflunine) every 3 weeks until disease progression or adverse effects became intolerable. The randomization included 266 patients, with 136 receiving erdafitinib and 130 receiving chemotherapy. The median age of all participants was 67 years, with 30% having undergone one prior line of therapy, 70% having undergone two lines of therapy, 74% having visceral metastases, and 90% having low PD-L1 expression (combined positive score < 10). The primary goal of this study was OS, with secondary objectives including PFS, response rate, and safety assessment.
After a median monitoring period of 15.9 months, erdafitinib was observed to enhance OS, decreasing death risk compared to chemotherapy, with a median survival of 12.1 months versus 7.8 months, a hazard ratio (HR) of 0.64, and a 95% confidence interval (CI) of 0.47–0.88, satisfying the predefined superiority criteria set by the Independent Data Monitoring Committee. The OS advantage offered by erdafitinib was consistent across various subgroups. Moreover, erdafitinib significantly extended the median PFS to 5.6 months from 2.7 months for chemotherapy, with an HR of 0.58 (95% CI, 0.44–0.78). The objective response rate was markedly higher with erdafitinib (45.6%) compared to chemotherapy (11.5%), with a relative risk of 3.94 (95% CI, 2.37–6.57; P < 0.001).
Both erdafitinib and chemotherapy demonstrated safety profiles consistent with previous findings. Significant treatment-related AEs occurred in 13.3% of the erdafitinib group and 24.1% of the chemotherapy group, with grade 3/4 events observed in 45.9% and 46.4% of the patients, respectively. Treatment-related fatalities were reported in one patient receiving erdafitinib and six patients undergoing chemotherapy. A higher rate of treatment-related dose reduction was noted for erdafitinib (66%) than for chemotherapy (21%), and 8% and 13% of patients discontinued treatment owing to AEs with erdafitinib and chemotherapy, respectively. Central serous retinopathy was diagnosed in 23 patients receiving erdafitinib (17%, grades 1–2 in 20 patients).
This evidence of the superiority of erdafitinib over chemotherapy, along with its safety profile demonstrated in the phase III THOR trial, led to the FDA approval of erdafitinib (Balversa; Janssen Biotech, Horsham Township, PA, USA) on January 19, 2024.52 This approval was obtained for patients with la/mUC harboring susceptible FGFR3 genetic alterations following progression after at least one prior line of systemic therapy. Erdafitinib is not advised for patients eligible for or yet to receive PD-1 or PD-L1 inhibitor therapy, amending its previously accelerated approval for patients with mUC with susceptible FGFR3 or FGFR2 alterations after platinum-containing chemotherapy.
Role of erdafitinib as combination therapy
A strong interest exists in combining FGFR inhibitors with immunotherapy to achieve synergy because FGFR inhibitors may help overcome the mechanism of resistance to immunotherapy presented by urothelial carcinomas with FGFR3 alterations.53 These tumors demonstrate a luminal-papillary molecular subtype and are often less immunogenic and lymphocyte-exclusionary.47
Erdafitinib, an FGFR inhibitor, inhibits driver oncogenes, releasing neoantigens and priming the immune system. Immunotherapy can then induce the clonal expansion of tumor-specific T-cell clones in the tumor microenvironment, leading to synergy.54
Luminal 1 subtype UC, which harbors FGFR3 mutations, lacks immune cell infiltration and has a lower response rate to ICIs.55 However, pre-clinical data from a genetically engineered murine model indicated that treatment with erdafitinib led to T-cell infiltration of the tumor and a decrease in Tregs, suggesting that FGFR-altered tumors could become more responsive to immunotherapy when combined with FGFR inhibitors.20
Prominent trials, including NORSE and FORT-2, have reported promising preliminary results.475657 The NORSE trial reported an ORR of 54.5% with a combination of erdafitinib and cetuximab, whereas the FORT-2 trial reported an ORR of 54% with rogaratinib combined with atezolizumab. These high response rates suggest the potential of this combination as a new standard of care, particularly for patients who are ineligible for upfront chemotherapy.
The FIERCE-22 trial and a study evaluating futibatinib with pembrolizumab demonstrated encouraging results, with an ORR of 30% and a safety profile that supports ongoing phase II studies.58 These trials enrolled patients with or without FGFR alterations, indicating the broad applicability of the combination approach.
The phase II NORSE trial evaluated the efficacy of erdafitinib in combination with the ICI cetrelimab in patients with mUC who were ineligible for cisplatin-based chemotherapy.55 The ORR and median PFS were better in the combination therapy group than those in the erdafitinib monotherapy group, supporting the effectiveness of this combination therapy.
The combination of erdafitinib and EV has been evaluated, highlighting the continuous exploration of FGFR inhibitors in combination with other therapies to improve outcomes in patients with mUC.59 These developments indicate a potential shift in the treatment paradigm for patients with FGFR-altered UC.
Table 1 summarizes notable clinical trials of FGFR inhibitors in mUC, focusing on patient populations and key findings.
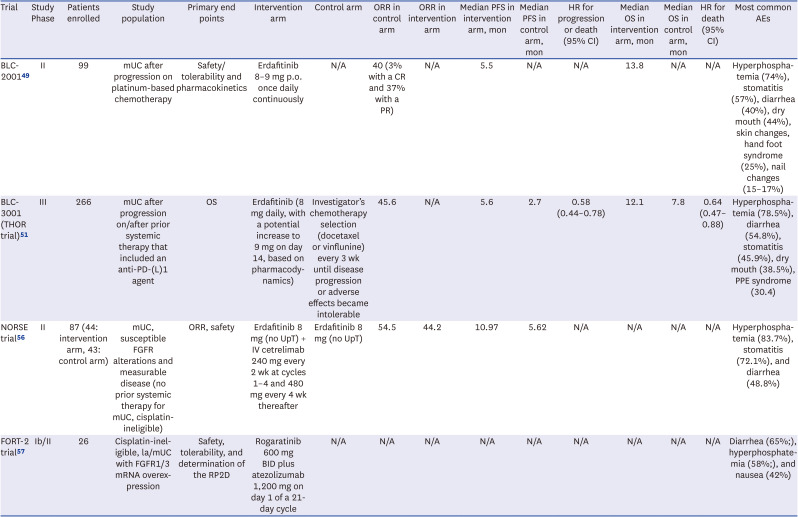
Table 1
Trials of FGFR inhibitors in urothelial cancer
| Trial | Study Phase | Patients enrolled | Study population | Primary end points | Intervention arm | Control arm | ORR in control arm | ORR in intervention arm | Median PFS in intervention arm, mon | Median PFS in control arm, mon | HR for progression or death (95% CI) | Median OS in intervention arm, mon | Median OS in control arm, mon | HR for death (95% CI) | Most common AEs |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BLC-200149 | II | 99 | mUC after progression on platinum-based chemotherapy | Safety/tolerability and pharmacokinetics | Erdafitinib 8–9 mg p.o. once daily continuously | N/A | 40 (3% with a CR and 37% with a PR) | N/A | 5.5 | N/A | N/A | 13.8 | N/A | N/A | Hyperphosphatemia (74%), stomatitis (57%), diarrhea (40%), dry mouth (44%), skin changes, hand foot syndrome (25%), nail changes (15–17%) |
| BLC-3001 (THOR trial)51 | III | 266 | mUC after progression on/after prior systemic therapy that included an anti-PD-(L)1 agent | OS | Erdafitinib (8 mg daily, with a potential increase to 9 mg on day 14, based on pharmacodynamics) | Investigator’s chemotherapy selection (docetaxel or vinflunine) every 3 wk until disease progression or adverse effects became intolerable | 45.6 | N/A | 5.6 | 2.7 | 0.58 (0.44–0.78) | 12.1 | 7.8 | 0.64 (0.47–0.88) | Hyperphosphatemia (78.5%), diarrhea (54.8%), stomatitis (45.9%), dry mouth (38.5%), PPE syndrome (30.4) |
| NORSE trial56 | II | 87 (44: intervention arm, 43: control arm) | mUC, susceptible FGFR alterations and measurable disease (no prior systemic therapy for mUC, cisplatin-ineligible) | ORR, safety | Erdafitinib 8 mg (no UpT) + IV cetrelimab 240 mg every 2 wk at cycles 1–4 and 480 mg every 4 wk thereafter | Erdafitinib 8 mg (no UpT) | 54.5 | 44.2 | 10.97 | 5.62 | N/A | N/A | N/A | N/A | Hyperphosphatemia (83.7%), stomatitis (72.1%), and diarrhea (48.8%) |
| FORT-2 trial57 | Ib/II | 26 | Cisplatin-ineligible, la/mUC with FGFR1/3 mRNA overexpression | Safety, tolerability, and determination of the RP2D | Rogaratinib 600 mg BID plus atezolizumab 1,200 mg on day 1 of a 21-day cycle | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | N/A | Diarrhea (65%;), hyperphosphatemia (58%;), and nausea (42%) |
ORR = objective response rate, PFS = progression-free survival, HR = hazard ratio, CI = confidence interval,
OS = overall survival, AE = adverse event, mUC = metastatic urothelial cancer, p.o. = per oral, N/A = not available, CR = complete response, PD-(L)1 = programmed death-(ligand) 1, PPE = palmar-plantar erythrodysesthesia, FGFR = fibroblast growth factor receptors, UpT =uptitration, IV = intravenous, la/mUC = locally advanced/metastatic urothelial cancer.
![]()
Rationale for FGFR targeting regardless of mutation status and specific clinical trials
FGFR inhibitors are primarily designed to target cancers with specific FGFR mutations, which lead to abnormal activation of the FGFR signaling pathway, a critical factor in the development of various cancers, including UC, breast cancer, and non-small cell lung cancer.32 However, FGFR inhibitors may still be effective even in the absence of detectable FGFR mutations due to several factors. First, the FGFR signaling pathway plays a role in regulating immune cells, angiogenesis, and epithelial-mesenchymal transition, all of which are essential components of the tumor microenvironment. By influencing these processes, FGFR inhibitors may impact tumor growth and progression indirectly, regardless of the presence of FGFR mutations.60 Additionally, some FGFR inhibitors, such as lenvatinib, exhibit broad targeting capabilities by also inhibiting other pathways or receptors, including the VEGFR and platelet-derived growth factor receptor family. This broad-spectrum inhibition can suppress cancer cell proliferation even when specific FGFR mutations are not present.61 Moreover, the effectiveness of FGFR inhibitors can be enhanced when used in combination with other treatments, such as ICIs.53 For example, the phase Ib/II FIERCE-22 (NCT03123055) trial evaluated the combination of the FGFR inhibitor vofatamab with pembrolizumab in previously treated mUC with FGFR alterations.62 The trial demonstrated an ORR of 40%, which is higher than the typically reported 20% response rate to ICIs alone. Notably, the trial showed similar response rates in both FGFR-altered (43%) and wild-type (40%) cohorts, suggesting that the combination of FGFR inhibition with ICIs may be effective regardless of the presence of FGFR mutations. This enhanced effectiveness may be due to FGFR inhibition increasing antigen expression and T-cell clonality, thereby making ICIs more effective.63 This approach is similar to the use of DNA damage response (DDR) gene targeting in prostate cancer. In prostate cancer, combining DDR inhibitors with androgen-targeting therapies has proven effective even in the absence of typical DDR mutations.6465 The synergy between these therapies, much like with FGFR inhibitors and ICIs, suggests that targeting multiple pathways can enhance therapeutic outcomes beyond the direct effects of the inhibitors alone.
Clinical evidence, such as the results from the FIERCE-22 trial, suggests the broader efficacy of FGFR inhibitors, indicating that these agents may offer therapeutic benefits beyond cases with identifiable FGFR mutations by potentially improving responses to other therapies, such as ICIs. While FGFR inhibitors are generally most effective in cancers with detectable FGFR mutations, their broader effects on the tumor microenvironment and other signaling pathways, along with evidence from studies like the FIERCE-22 trial and parallels with DDR targeting strategies in prostate cancer, suggest that they could still provide therapeutic benefits even in the absence of such mutations.
Go to :
DIFFERENCES IN FGFR MUTATION IN UC BETWEEN ASIANS (INCLUDING KOREANS) AND WESTERNERS
A recent study conducted at a single center in Korea explored the prevalence and clinical significance of FGFR3 mutations in UC using next-generation sequencing.66 The research included 123 patients treated at Chonnam National University Hospital, covering various forms of UC. The study found that FGFR3 mutations occurred in 30.1% of the patients, with notable variation across UC types. The highest mutation rate was detected in NMIBC, affecting 45.1% of patients, followed by 22.7% in MIBC and 14.3% in upper tract urothelial carcinoma (UTUC). These mutations were predominantly associated with early-stage disease and a heightened risk of recurrence in NMIBC. When these results are compared with those from Western populations, a clear pattern of similarity emerges. Both Korean and Western studies indicate that FGFR3 mutations are most common in NMIBC, with Western research reporting mutation rates between 49% and 84%, comparable to the 45.1% observed in the Korean cohort. The lower mutation rates in MIBC and UTUC found in the Korean study, 22.7% and 14.3% respectively, also align with data from Western populations.3067686970 On the other hand, a study comparing Western and Chinese UC cases revealed a significantly lower frequency of FGFR3 mutations among Han Chinese patients, with rates of 9.4% in BC, 8.8% in renal pelvic carcinoma, and 2.6% in ureter carcinoma, which are notably lower than those reported in Western populations.71 Additionally, research involving a large cohort of patients with UC identified FGFR3 mutations in 17.4% of cases, with a higher prevalence observed among individuals of European ancestry compared to other ethnicities.72
Several studies have examined the differences in treatment outcomes related to FGFR mutations between Asian and Western populations in UC. The MONSTAR-SCREEN database study conducted in Japan reported that FGFR2/3 genetic alterations were present in 11.6% of advanced/metastatic UC cases. However, PFS did not significantly differ between FGFR-positive and FGFR-negative patients, highlighting a unique response pattern in the Japanese cohort.73 Additionally, an Asian subgroup analysis of the THOR phase III study revealed that erdafitinib significantly improved survival outcomes in Asian patients with FGFR alterations, showing better efficacy compared to chemotherapy, which aligns with global data but highlights specific benefits within the Asian population.74
Previous studies have shown varying results, particularly when comparing Asian and Western populations, which highlights the need for further research to validate these findings across different ethnic groups. Future studies should focus on confirming these results and evaluating the clinical applications of FGFR3 as a biomarker, taking into consideration the potential differences between Asian and Western populations.
Go to :
MECHANISMS OF RESISTANCE
De novo FGFR alterations typically promote tumorigenesis, whereas additional mutations that enable survival and evolution under certain selection pressures can be acquired.75 Resistance to FGFR inhibitors can reflect the presence of additional somatic FGFR mutations, including innate gatekeeper mutations in some primary tumors and acquired resistance mutations in post-treatment tumors.76 Analysis of serial biopsy, cfDNA, and circulating tumor DNA samples indicates that on-target resistance mutations are frequently polyclonal, and multiple mutations can be acquired by the same patient during treatment.77 Secondary mutations occur preferentially in the tyrosine kinase domain, thereby interfering with the ability of FGFR inhibitors to access drug-binding clefts.78
Several point mutations in FGFR2, including V564F/I/L, E565A, and L617F/M/V, preferentially occur in patients with cholangiocarcinoma (CCA) with disease progression on FGFR-targeted therapies; however, other FGFR2 kinase domain mutations have been detected in pan-cancer cohorts.75 The clinical benefits of FGFR inhibitors can be compromised by several types of resistance mutations, including gatekeeper, molecular brake, and DFG-latch mutations.76 Preliminary data from clinical trials suggest that third-generation selective FGFR2 or FGFR3 inhibitors can overcome many resistance mutations associated with earlier-generation agents, thus providing more durable clinical benefits.79
Bypass signaling provides alternative mechanisms of resistance to FGFR inhibitors. The activation of other RTKs and downstream signaling cascades can compensate for the inhibition of FGFR signaling.16 The epidermal growth factor receptor (EGFR) and mesenchymal epithelial transition are examples of RTKs that can activate these cascades.80 Concurrent alterations, especially the amplification of genes encoding RTKs or their downstream signaling components or targets, are sources of intrinsic resistance to FGFR inhibitors.81 Pretreatment cfDNA sequencing of plasma samples revealed alterations in EGFR, BRAF, and CCND1 in patients with mUC harboring FGFR2/3 alterations.82
TP53 alterations typically elicit defective sensing of and response to DNA damage, contributing to tumor progression and potentially context-dependent drug resistance.83 FGFR2 alterations and TP53 mutations occur in distinct CCA subtypes, and their co-occurrence can influence the clinical outcomes of FGFR inhibitor therapy.84 Concurrent TP53 mutations are frequent in many other cancer types and may induce resistance to FGFR inhibitors via the activation of RTK signaling.75
A systematic analysis of data from studies involving patients with FGFR2-altered intrahepatic CCA and disease progression treated with FGFR inhibitors reported the occurrence of secondary mutations in the FGFR2 kinase domain.27 Longitudinal genomic studies provide evidence that the on-target mechanisms underlying acquired resistance to FGFR inhibitors are complex and that resistance can arise from either single or multiple acquired mutations in the kinase domains of FGFRs.75 Ongoing clinical trials and future research are required to obtain a comprehensive genomic landscape of acquired resistance to FGFR inhibitors and optimize clinical benefits through the development of next-generation inhibitors.
Go to :
FUTURE PERSPECTIVES ON FGFR INHIBITORS
The role of FGFR as a therapeutic target in mUC is well-established, suggesting that ongoing research could further elucidate its significance in NMIBC and MIBC as well. Given the promising results of erdafitinib in addressing FGFR-altered UC, numerous clinical trials are actively investigating its application in various treatment scenarios. One key study, the THOR-2 trial (NCT04172675) evaluated erdafitinib in high-risk NMIBC patients with FGFR3/2 alterations who had recurrent disease after Bacillus Calmette-Guérin (BCG) treatment and were ineligible for or refused cystectomy.85 The results showed that erdafitinib significantly reduced the risk of recurrence or death by 72% compared to chemotherapy. However, 31% of patients experienced severe AEs, leading to a 29% discontinuation rate. Additionally, the exploration of erdafitinib delivery through an intravesical system named TAR-210 is underway for individuals with specific FGFR mutations and fusions.55 This study will include a dose escalation phase for eligible participants who have previously received BCG treatment for recurrent high-risk papillary NMIBC and are either unable or unwilling to undergo cystectomy. The subsequent part of the study will expand the doses to two groups: those with NMIBC and those with MIBC, as outlined in the ClinicalTrials.gov identifier NCT05316155.86
Further investigations include the NERA trial, which is focused on the efficacy of erdafitinib in treating patients with MIBC with particular FGFR2/3 alterations who cannot receive cisplatin-based chemotherapy.55 This trial, registered under the ClinicalTrials.gov identifier NCT05564416, will measure its primary outcome based on pathologic complete response rates at cystectomy by administering erdafitinib either alone or alongside atezolizumab. Moreover, a phase Ib trial, identified by ClinicalTrials.gov identifier NCT04963153, is assessing the combined use of erdafitinib and EV in patients with FGFR2/3-altered mUC who do not respond to chemotherapy and immunotherapy, focusing on the treatment's feasibility, safety, maximum tolerated dose, and recommended phase 2 dose.55
However, challenges such as treatment duration and systemic side effects associated with erdafitinib therapy remain concerns. Additionally, the issue of acquired resistance to erdafitinib and other TKIs that target the FGFR signaling pathway is a significant clinical hurdle. Resistance mechanisms often involve compensatory signaling pathways such as the PI3K pathway, which has been identified as a mechanism of resistance to the FGFR inhibitor AZD4547.87 Strategies to overcome resistance, including the development of new-generation therapies and combination therapies targeting both EGFR and FGFR, are under consideration.88
The scientific community has worked diligently to translate the preclinical success of FGFR inhibitors into clinical practice. Despite challenges such as target selectivity and AEs, erdafitinib received FDA approval in April 2019 for cisplatin-refractory FGFR2/FGFR3-altered BC, following the BCL2001 phase II clinical trial results.89 The THOR phase III trial results could further clarify the role of erdafitinib in Europe.90
The combination of FGFR inhibitors with other therapies presents a promising developmental direction, as exemplified by the NORSE phase I/II trial and other ongoing studies assessing the efficacy and safety of such combinations in advanced or metastatic FGFR-altered UC.55 Early-stage intervention trials are in progress to evaluate erdafitinib alone or in combination with other drugs for NMIBC or MIBC candidates for adjuvant treatment.9192 Among these, the PROOF-302 phase III trial is focused on infigratinib as an adjuvant therapy for cisplatin-ineligible patients with MIBC harboring FGFR3 alterations, alongside other studies aimed at addressing the effectiveness of FGFR inhibitors in earlier disease stages.92 However, challenges such as patient enrollment have led to the discontinuation of certain developmental efforts, highlighting the complex landscape of FGFR inhibitor research and development. Ongoing trials with selective FGFR inhibitors including the trials described above are summarized in Table 2.
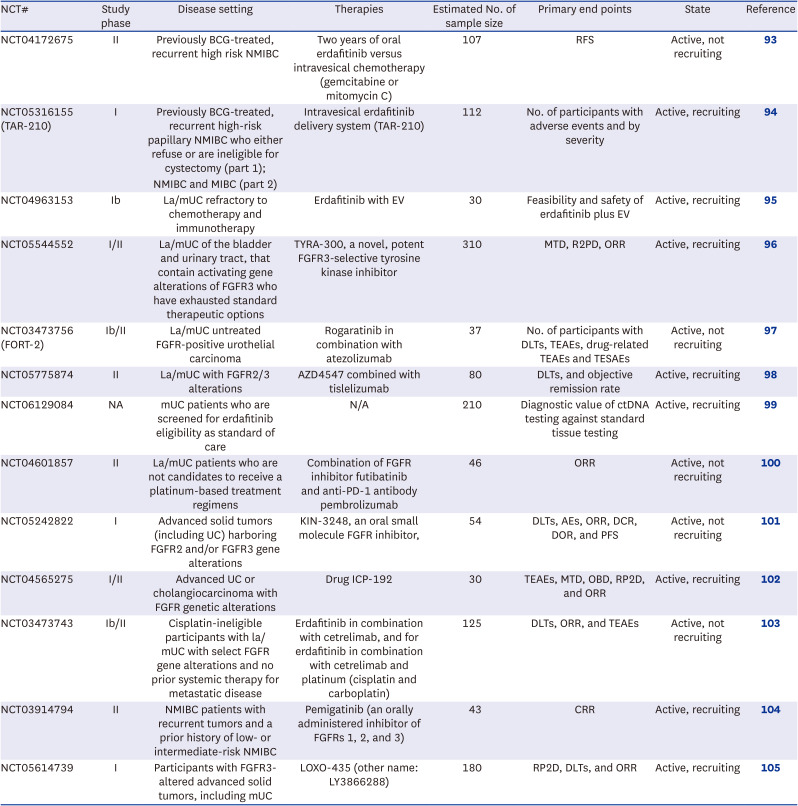
Table 2
Ongoing clinical trials evaluating FGFR inhibitors in urothelial cancer
| NCT# | Study phase | Disease setting | Therapies | Estimated No. of sample size | Primary end points | State | Reference |
|---|---|---|---|---|---|---|---|
| NCT04172675 | II | Previously BCG-treated, recurrent high risk NMIBC | Two years of oral erdafitinib versus intravesical chemotherapy (gemcitabine or mitomycin C) | 107 | RFS | Active, not recruiting | 93 |
| NCT05316155 (TAR-210) | I | Previously BCG-treated, recurrent high-risk papillary NMIBC who either refuse or are ineligible for cystectomy (part 1); | Intravesical erdafitinib delivery system (TAR-210) | 112 | No. of participants with adverse events and by severity | Active, recruiting | 94 |
| NMIBC and MIBC (part 2) | |||||||
| NCT04963153 | Ib | La/mUC refractory to chemotherapy and immunotherapy | Erdafitinib with EV | 30 | Feasibility and safety of erdafitinib plus EV | Active, recruiting | 95 |
| NCT05544552 | I/II | La/mUC of the bladder and urinary tract, that contain activating gene alterations of FGFR3 who have exhausted standard therapeutic options | TYRA-300, a novel, potent FGFR3-selective tyrosine kinase inhibitor | 310 | MTD, R2PD, ORR | Active, recruiting | 96 |
| NCT03473756 (FORT-2) | Ib/II | La/mUC untreated FGFR-positive urothelial carcinoma | Rogaratinib in combination with atezolizumab | 37 | No. of participants with DLTs, TEAEs, drug-related TEAEs and TESAEs | Active, not recruiting | 97 |
| NCT05775874 | II | La/mUC with FGFR2/3 alterations | AZD4547 combined with tislelizumab | 80 | DLTs, and objective remission rate | Active, recruiting | 98 |
| NCT06129084 | NA | mUC patients who are screened for erdafitinib eligibility as standard of care | N/A | 210 | Diagnostic value of ctDNA testing against standard tissue testing | Active, recruiting | 99 |
| NCT04601857 | II | La/mUC patients who are not candidates to receive a platinum-based treatment regimens | Combination of FGFR inhibitor futibatinib and anti-PD-1 antibody pembrolizumab | 46 | ORR | Active, not recruiting | 100 |
| NCT05242822 | I | Advanced solid tumors (including UC) harboring FGFR2 and/or FGFR3 gene alterations | KIN-3248, an oral small molecule FGFR inhibitor, | 54 | DLTs, AEs, ORR, DCR, DOR, and PFS | Active, not recruiting | 101 |
| NCT04565275 | I/II | Advanced UC or cholangiocarcinoma with FGFR genetic alterations | Drug ICP-192 | 30 | TEAEs, MTD, OBD, RP2D, and ORR | Active, recruiting | 102 |
| NCT03473743 | Ib/II | Cisplatin-ineligible participants with la/mUC with select FGFR gene alterations and no prior systemic therapy for metastatic disease | Erdafitinib in combination with cetrelimab, and for erdafitinib in combination with cetrelimab and platinum (cisplatin and carboplatin) | 125 | DLTs, ORR, and TEAEs | Active, not recruiting | 103 |
| NCT03914794 | II | NMIBC patients with recurrent tumors and a prior history of low- or intermediate-risk NMIBC | Pemigatinib (an orally administered inhibitor of FGFRs 1, 2, and 3) | 43 | CRR | Active, recruiting | 104 |
| NCT05614739 | I | Participants with FGFR3-altered advanced solid tumors, including mUC | LOXO-435 (other name: LY3866288) | 180 | RP2D, DLTs, and ORR | Active, recruiting | 105 |
FGFR = fibroblast growth factor receptor, NCT = National Clinical Trial, BCG = Bacillus Calmette-Guérin, NMIBC = non-muscle-invasive bladder cancer, MIBC = muscle-invasive bladder cancer, la/mUC = locally advanced/metastatic urothelial cancer, EV = enfortumab vedotin, MTD = maximum tolerated dose, RP2D = recommended phase 2 dose, ORR = objective response rate, DLT = dose-limiting toxicity, TEAE = treatment-emergent adverse event, TESAE = treatment-emergent serious adverse event, mUC = metastatic urothelial cancer, N/A = not available, ctDNA = circulating tumor DNA, PD-1 = programmed death-1, UC = urothelial cancer, AE = adverse event, DCR = disease control rate, DOR = duration of response, PFS = progression-free survival, OBD = optimal biological dose, CRR = complete response rate.
![]()
Go to :
CONCLUSION
The FGF/FGFR axis plays a critical role in the development and progression of BC, particularly through FGFR3 mutations that significantly promote tumor growth. Traditionally, platinum-based chemotherapy has been the mainstay of BC treatment. However, the introduction of targeted anti-FGFR therapies represents substantial progress in the treatment of patients with specific FGFR mutations. Initially, these FGFR-targeted treatments faced challenges owing to their broad activity spectrum, which led to adverse effects by impacting multiple receptors. Recent advancements have led to the development of a more selective FGFR inhibitor, erdafitinib, which received FDA approval for treating advanced BC with FGFR alterations after platinum-based therapy failure, based on the BLC2001 trial results. This marks a significant shift in the treatment landscape, offering more nuanced options; however, it introduces complexity as more FGFR inhibitors are being studied.
The utility of these treatments, especially in patients with BC harboring FGFR3 mutations, is currently under close observation. Typically, therapies such as erdafitinib are considered after the failure of cisplatin and immunotherapy. However, the response of FGFR-altered BC to these treatments varies, indicating the need for further investigation into the sequencing of treatments and the impact of prior immunotherapy on outcomes. Emerging therapies, such as EV and sacituzumab govitecan, can provide additional options for patients with FGFR-altered, cisplatin- and immunotherapy-refractory, underscoring the importance of understanding resistance mechanisms to FGFR inhibitors for developing effective combination strategies.
The approval of erdafitinib as the only FGFR inhibitor for specific mutations in UC highlights the beginning of a new treatment era, necessitating a multidisciplinary approach to FGFR testing, toxicity management, and patient education. Ongoing research on alternative diagnostic methods and the optimal sequencing of new systemic therapies is crucial. Future clinical trials should aim to investigate the potential of other FGFR inhibitors, their use in the earlier stages of the disease, and various combination treatments, emphasizing the need for further studies to refine the clinical application of FGFR inhibitors and enhance patient care for UC.
Go to :
 XML Download
XML Download