

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
It’s been over 200 years since James Parkinson published a paper titled “Shaking Palsy.”1,2 While the first 100 years leading up to the discovery of Lewy bodies were marked by little change, the next 100 years have seen remarkable progress.3 Various medications, including levodopa, dopa agonists, Monoamine Oxidase B (MAO-B) inhibitors, Catechol-O-Methyltransferase (COMT) inhibitors, amantadine, anticholinergics, etc., have been developed and used to treat the symptoms of the disease, and new techniques, such as deep brain stimulation and levodopa-carbidopa intestinal gel, for advanced cases, have been developed and used. There have also been remarkable advances in diagnosis, including the development of dopamine transporter (DAT) imaging and magnetic resonance imaging techniques that can reflect levodopa-responsive parkinsonism, a core feature of Parkinson’s disease (PD).4-7
On the other hand, research into the cause of the disease has been slower to develop.3 Although the discovery of the SNCA gene and various experimental and clinical studies have shown that alpha-synuclein (AS) is essential in the pathogenesis of the disease, it is difficult to explain the cause easily because various other pathogenic mechanisms are involved.8 This is a major hurdle in discovering disease-modifying therapies (DMTs). This is because most drugs target only one or two problems in the pathogenesis, so temporarily blocking one is unlikely to stop the progression of the disease. Nonetheless, it is hoped that recent advances like seeding amplification assays in detecting AS in biofluids and tissues of PD patients and recent successes in treating Alzheimer’s disease will lead to therapies that address the disease progression itself.9-11
PD is known to have a long prodromal period.12 In particular, non-motor symptoms appear as early as a few years and as long as 15 to 20 years before motor symptoms become evident. This is also related to the pathologic location of AS, which is believed to cause the disease.13 For example, lesions of vagal nucleus in the medulla oblongata is known to be related to constipation in the prodromal phase. Also, lesions lower raphe nucleus and locus coeruleus in the pons could result in depression and rapid eye movement sleep behavior disorder (RBD), respectively. Based on these findings, efforts are being made to find earlier and earlier stages.12 This is necessary for the early application of DMTs, which are expected to be developed in the near future, even if there is no immediate method. This review will discuss related studies centered on the body-first and brain-first theory, which are based on recent studies regarding the starting point of pathological changes in PD.
PARKINSON’s DISEASE AND GUT
The gut is very important in PD. First of all, symptoms related to the gastrointestinal (GI) tract are common in PD. Drooling, although associated with decreased swallowing, has been reported in 10% to 81% of PD patients.14 More importantly, gastroparesis is reported to have a prevalence of 70-100% in the majority of PD patients. Constipation, a lower GI symptom, is just as common as gastroparesis, occurring in 80-90% of PD patients. Although constipation is nonspecific, it is considered an essential prodromal non-motor symptom because it precedes motor symptoms.12
How do these GI symptoms affect PD patients? Basically, they limit their food intake. Indigestion is expected due to slow bowel movements. To make matters worse, antiparkinsonian medications with dopaminergic property make bowel movements slower.15 The converted dopamine from levodopa also stimulates the chemoreceptor trigger zone.16,17 Thus, vomiting is common with medications. In addition, slow bowel movement interferes with the absorption of levodopa, making it difficult to see its effectiveness.18 This is especially common in advanced PD, where the drug does not reach the jejunum quickly enough to be absorbed and is lost in the middle, causing delayed or partial ON state.19-21
In PD, the gut doesn’t just affect quality of life or motor fluctuations but also provides clues to the pathogenesis of the disease. The synucleinopathy in the GI tract of PD patients was reported in 1980s.22,23 Later, Braak et al.24 reported again that synuclein pathologies were deposited in the submucosal plexus and myenteric plexus in PD patients, suggesting they may be transported to the central nervous system via the vagus nerve.
FROM GUT TO BRAIN
For the hypothesis that synucleinopathy originates in the gut and is transported to the brain to be proven,24 several supportive pieces of evidence are needed. First, synucleinopathy should be more evident in the GI tract of PD patients compared to non-Parkinson’s controls. Most studies since Braak et al.24 report have shown GI pathology in people with PD.25 In addition, the vagus nerve should be able to act as a conduit for AS delivery. This has already been shown in animal studies.26,27 When AS preformed fibrils and patient-derived AS aggregates were injected into the GI tract and delivered to the brain via the vagus nerve. Finally, it should be demonstrated that cutting the vagus nerve does not transmit pathologic changes. Animal studies have shown that vagotomy after GI infusion of pathologic AS does not result in exogenous AS-induced pathologic changes in the brain,28 and clinical studies that have followed vagotomy patients for years have shown a lower incidence of PD in the group that underwent truncal vagotomy.29,30
BRAIN-FIRST VERSUS BODY-FIRST
RBD is the prodromal symptom with the highest relative risk of developing PD among all prodromal symptoms.12 Therefore, finding which body areas are involved in patients with RBD without clinical parkinsonism may help to understand the progression pattern of the disease. One study compared DAT scans and metaiodobenzylguanidine (MIBG) myocardial scintigraphy in patients with RBD and autonomic dysfunction.31 Of the 18 patients in the study, 56% (10 patients) had positive DAT scans, while 94% (17 patients) had positive MIBG myocardial scintigraphy. The anatomical location suggests that the heart’s autonomic nerves were already affected before the dopamine cell’s death in the substantia nigra, which is responsible for motor symptoms, suggesting that the pathology may have started in the body and progressed to the brain.
What about PD patients without RBD? Given the spreading property of AS pathologic changes, PD patients without RBD may have a different starting point of pathology than those with RBD. Some studies have performed MIBG myocardial scans and donepezil positron emission tomography/ computed tomography (PET/CT) in idiopathic RBD subjects, PD patients with RBD, and PD patients without RBD to confirm this.32,33 As expected, MIBG myocardial scans were abnormal in most patients with RBD, regardless of PD status, and relatively normal in PD patients without RBD. Donepezil PET/CT, which reflects bowel autonomic function related to motility, showed a similar pattern to the MIBG myocardial scan results. Eventually, the presence of RBD is deeply correlated with the autonomic nervous system involvement of the body.
ALPHA-SYNUCLEIN ORIGIN SITE AND CONNECTOME (SOC) MODEL
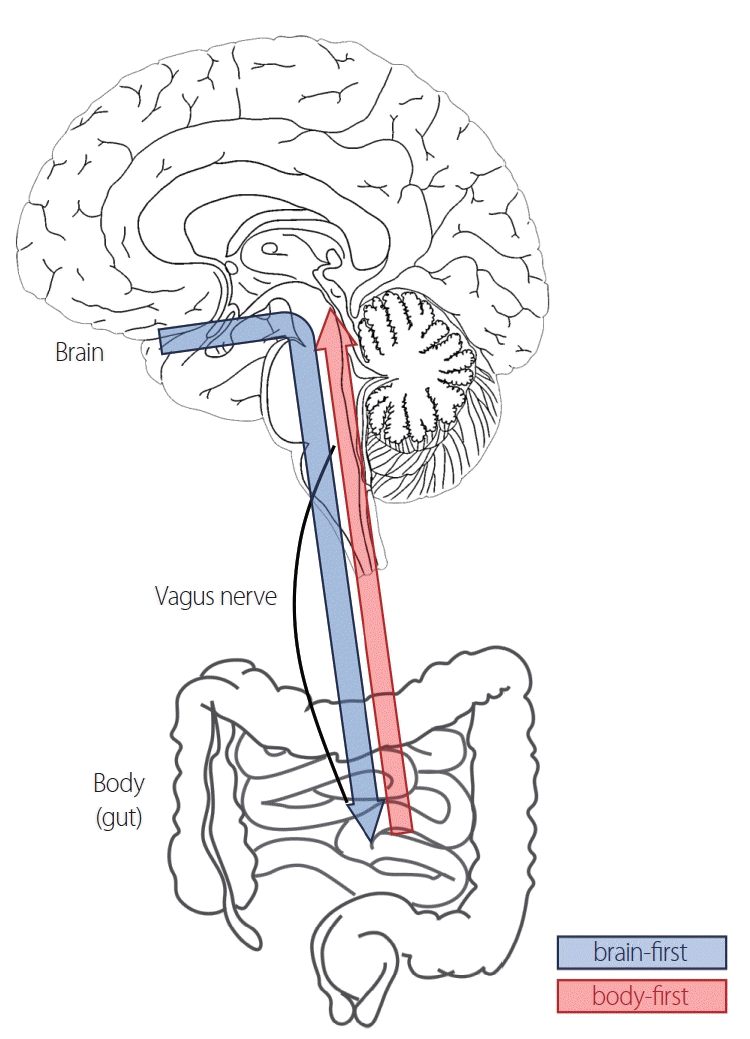
The brain-first versus body-first proposal advances to the SOC model.32,34 The idea is that there is one location where AS pathology initially begins and that the pathological changes spread through the connectome to vulnerable parts of the nervous system. Thus, if the pathology originates in the brain, it would be a brain-first subtype (the olfactory bulb [OB] or amygdala would be the primary starting point within the brain) (Fig. 1). On the other hand, if the pathology originates in the body, it is thought that the most likely origin is the enteric nervous system and affects the heart and brainstem via the peripheral autonomic nervous system. The latter case is the type that is well represented in Braak’s staging.35
Since the authors assume that the pathological changes occur in a single location, they can think of brain-first PD as spreading along the ipsilateral connectome from a single location.34 This helps explain the asymmetry of symptoms and DAT scans in the brain-first type. In contrast, body-first PD starts in the enteric system, so by the time it affects the brainstem via the vagus nerve, it already has a symmetric distribution. Therefore, it is predictable that the DAT scan abnormalities and clinical symptoms will be more symmetric. They also found that the two types showed different patterns in the distribution of pathology: the amygdala-predominant type almost always involved the OB, with less involvement of the medulla or pons.34-37 In contrast, the brainstem-predominant type had very little involvement in the OB.
Clinically, the SOC model can explain a lot. In body-first PD, non-motor symptoms such as RBD, depression, autonomic dysfunction, and constipation are common in the prodromal phase, and the high symmetry of motor symptoms can be well explained. On the other hand, in the brain-first type, prodromal symptoms are rarely found, and the asymmetry of motor symptoms can be well understood.
CONTROVERSY ON THE SOC MODEL
However, not everything about this model is well acceptable. Many clinical studies in PD have shown that RBD, autonomic dysfunction, hyposmia, rapid progression, faster cognitive impairment, and faster disease progression are linked.38-40 In the SOC model, RBD and early autonomic dysfunction are the main symptoms of body-first PD. So, the authors of the SOC model also grouped them together as symptoms of the body-first type.34
However, this raises a few questions. First, in the body-first type of pathology, the OB is not often found to have pathology, but rather, the degree of hyposmia is more severe.34 The authors explain that hyposmia is a kind of sensation that patients feel, and what affects olfaction is not only the OB but also the olfactory cortex and memory that process it. They also speculate that hyposmia is less likely to occur in brain-first PD because OB involvement is more often unilaterally involved in this type. However, evidence is still lacking on this point. It should also be noted that hyposmia is a relatively early symptom,41 so it is difficult to imagine that cortical involvement would influence more body-first PD.
The second issue is that cognitive decline is faster in the body-first type. This is also unexpected because it seems that a disease that starts in the brain should progress faster, but it does not.34 The authors explain that in the case of body-first PD, the overall AS burden has already accumulated in the cortex as the disease progresses slowly over a long period. In other words, even if motor symptoms have appeared just before, different brain areas already have been involved to some extent in body-first PD. The brain-first type, on the other hand, starts in the amygdala or OB on one side of the brain and is predominantly symptomatic on that side, and the disease must be quite advanced before it moves to the other side. However, even in this case, there seems to be a disadvantage in that it is difficult to explain the lack of severe dementia symptoms when the disease spreads heavily on one side and the reason for the slow progression to the contralateral side within the brain.42
The third point is the tendency to dichotomize clinical symptoms overly. For instance, while asymmetry may appear more frequently in young-onset Parkinson’s disease, it is still present in 92.3% of older patients.43 Therefore, asymmetry cannot be considered a hallmark feature of young-onset PD. As a result, linking asymmetry to other characteristics of young-onset PD, such as slower progression or genetic predisposition, presents significant challenges. Some studies showed that asymmetry is not associated with prognosis.44 Further studies are necessary to support a clinical association between asymmetry and the other clinical features of the brain-first type.
Also, exceptions have been observed in which patients with RBD do not exhibit evidence of body involvement on MIBG scans. These findings could stem from the limitations of current imaging techniques in detecting mild neurodegeneration by synucleinopathies. Moreover, cut-off values have not yet been established across diverse age ranges or ethnic groups due to the limited data available on MIBG or DAT imaging.
Fearon et al.45 expressed concerns that this model might artificially dichotomize the PD clinical type based on the phenomenon observed in the bidirectional extension of AS in animal models. They pointed out that the assumption that the pathology originates from a single point and is propagated through neural connections could not reflect the heterogeneity of PD.34 For example, they noted that RBD is a symptom caused by the involvement of the pons, and it is difficult to explain this given the various studies showing that extensive parts of the brain and brainstem are already affected by that time. Additionally, they highlighted that the average age of onset in body-first PD is about 10 years older than in brain-first PD, suggesting that the comparison is unfair. Moreover, since there is no longitudinal data, they raised the possibility that classification might change over time, similar to how the tremor-dominant type might be reclassified as the akinetic-rigid type. The fact that most PD patients have copathologies also supports the notion that a single consistent pattern of AS transmission may not fully explain the disease.46,47
CONCLUSION
PD is a unique condition. This is because dopaminergic cell degeneration is clearly visible on a DAT scan, and this degeneration is closely associated with a pronounced response to medication. However, aside from this, the other symptoms vary greatly from person to person. While many patients experience prodromal phase symptoms such as RBD, constipation, and depression, there are also many who do not. Neurologically, symptoms reflect localization, so the location of the lesion can be predicted based on the symptoms. However, it is difficult to determine when these changes began. Although there is some overlap between the initiation of pathological changes, functional problems, and cell death, these events do not necessarily coincide.
The intriguing proposal of brain-first versus body-first PD has emerged through the integration of previously known pathological findings, newly acquired knowledge from advances in functional imaging, and the clustering of clinical symptoms.32-34 While this proposal warrants repeated studies including longitudinal observations and continued discussion, it must also be reviewed critically and objectively. Hopefully, with the future development of more precise imaging and other biomarker techniques that reflect in-vivo pathology, this controversy can be resolved, leading to significant discoveries that will aid in selecting patients for DMTs.
 XML Download
XML Download