An 8-year-old girl presented to the emergency department of Seoul National University Children’s Hospital with a 4-day history of fever and gross hematuria, accompanied by periorbital edema that developed on the day of the visit. She had a congenital single kidney and a history of epilepsy following encephalitis at the age of 2 years. Her family history included a maternal uncle who underwent kidney transplantation due to glomerulonephritis, and her mother and older sister experienced recurrent hematuria of unknown causes. However, there was no family history of autoimmune diseases, including SLE. Laboratory findings revealed profound proteinuria (urine protein-to-creatinine ratio [UPCR] 11.6 mg/mg) with hypoalbuminemia (2.7 g/dL), consistent with nephrotic syndrome, and red blood cell >100 per high power field in urinalysis, which suggested glomerulonephritis with mildly decreased kidney function (
Table 1). The antinuclear antibody (ANA) was positive with a homogeneous pattern, along with marginally low to normal complement levels (C3/C4 82/10 mg/dL), slightly elevated anti-double stranded DNA (anti-dsDNA) antibodies (21.3 IU/mL), and positive LA; the complete blood count was normal (
Tables 1,
2). She had no clinical symptoms of SLE, such as rash, arthralgia, or alopecia. Kidney biopsy revealed IgAN (Oxford classification M1 S0 E1 T0) with an MPGN pattern, which was characterized by diffuse moderate hypercellularity of mesangial and endothelial cells, tram-track appearance, and moderate mesangial and subendothelial deposits with predominant IgA (3+) (
Fig. 1). Oral glucocorticoid (GC) therapy with deflazacort 60 mg/m
2/day was initiated for 1 month; however, due to persistent nephrotic-range proteinuria with periorbital edema, mycophenolate mofetil (MMF) 400 mg/m
2/dose twice daily was added, with a gradual tapering of deflazacort. Two months after diagnosis, she presented to the emergency department because of left upper extremity swelling. Imaging studies, including upper extremity Doppler ultrasound and computed tomography angiography, revealed multiple thromboses in the left axillary, subclavian, and brachial veins (
Fig. 2). She was treated with intravenous heparinization followed by 4 months of oral warfarin, which resolved the thrombosis. The coagulation panel and tests for factors IX to XII to investigate the cause of the thrombosis were normal. At that time, SLE was suspected, and relevant tests were conducted. The results showed positive results for ANA and LA; however, complement levels, anti-dsDNA, anti-Smith, anti-cardiolipin, and anti-β2GPI antibodies were within normal ranges; therefore, she did not meet the diagnostic criteria for APS or SLE (
Table 2). While on continued treatment with deflazacort (60 mg/m
2/day for 1 month, then tapered) and MMF (increased from 400 mg/m
2/dose to 600 mg/m
2/dose, twice daily), proteinuria achieved and maintained in remission, allowing for gradual tapering and eventual discontinuation of immunosuppressants (deflazacort at 6.5 months and MMF at 7 months). Thereafter, she was maintained on enalapril (0.1 mg/kg/day) alone. However, 10 months after stopping the immunosuppressants, she experienced gross hematuria recurrence, purpura on both legs, and recurrent epistaxis. Laboratory findings showed prolonged prothrombin time (PT) (international normalized ratio, 1.56) and activated partial thromboplastin time (aPTT) (69.3 seconds), hypocomplementemia (C3/C4, 45/2 mg/dL), acute kidney injury, and worsened proteinuria (UPCR, 2.3 mg/mg) (
Table 1). Further workup for suspected SLE and LN revealed positive ANA (>1: 320), high anti-dsDNA antibody titer (907.5 IU/mL), and triple aPL positivity (
Table 2). Factor assays revealed factor II deficiency (41%; reference range, 70%–120%), whereas the other factors were normal. At this point, the initial diagnosis of IgAN was reclassified, as the patient fulfilled the 2019 European League Against Rheumatism/American College of Rheumatology (EULAR/ACR) criteria for SLE, with a score of 22, leading to a diagnosis of LN, along with APS and LAHPS (
Table 3). Treatment with intravenous methylprednisolone pulse (30 mg/kg/dose for 3 days), followed by oral GC (prednisolone 2 mg/kg/day) and MMF (600 mg/m
2/dose twice daily) led to remission of proteinuria. GC was discontinued after 2 years, with MMF being continued. However, 2 years after cessation of GC, proteinuria worsened and did not improve despite reintroducing GC (prednisolone 1 mg/kg/day) and increasing the MMF dose up to 800 mg/m
2/dose twice daily. Rituximab (375 mg/m
2/dose, weekly, total 4 doses) induced an initial response, but nephrotic-range proteinuria relapsed. She is currently receiving monthly cyclophosphamide (500 mg/m
2/dose) in addition to GC and MMF. Despite the introduction of cyclophosphamide, nephrotic-range of proteinuria (UPCR, 12.24 mg/mg) has persisted, along with a mild decrease in kidney function (estimated glomerular filtration rate 83.0 mL/min/1.73 m
2, calculated by the Schwartz equation 2012). However, the SLE activity markers have improved, recently normalizing (anti-dsDNA antibodies <0.1 IU/mL and C3/C4 125/19 mg/dL) at the last follow-up at age 15 years (
Tables 1, 2). Additionally, no further thrombotic or bleeding events have occurred.
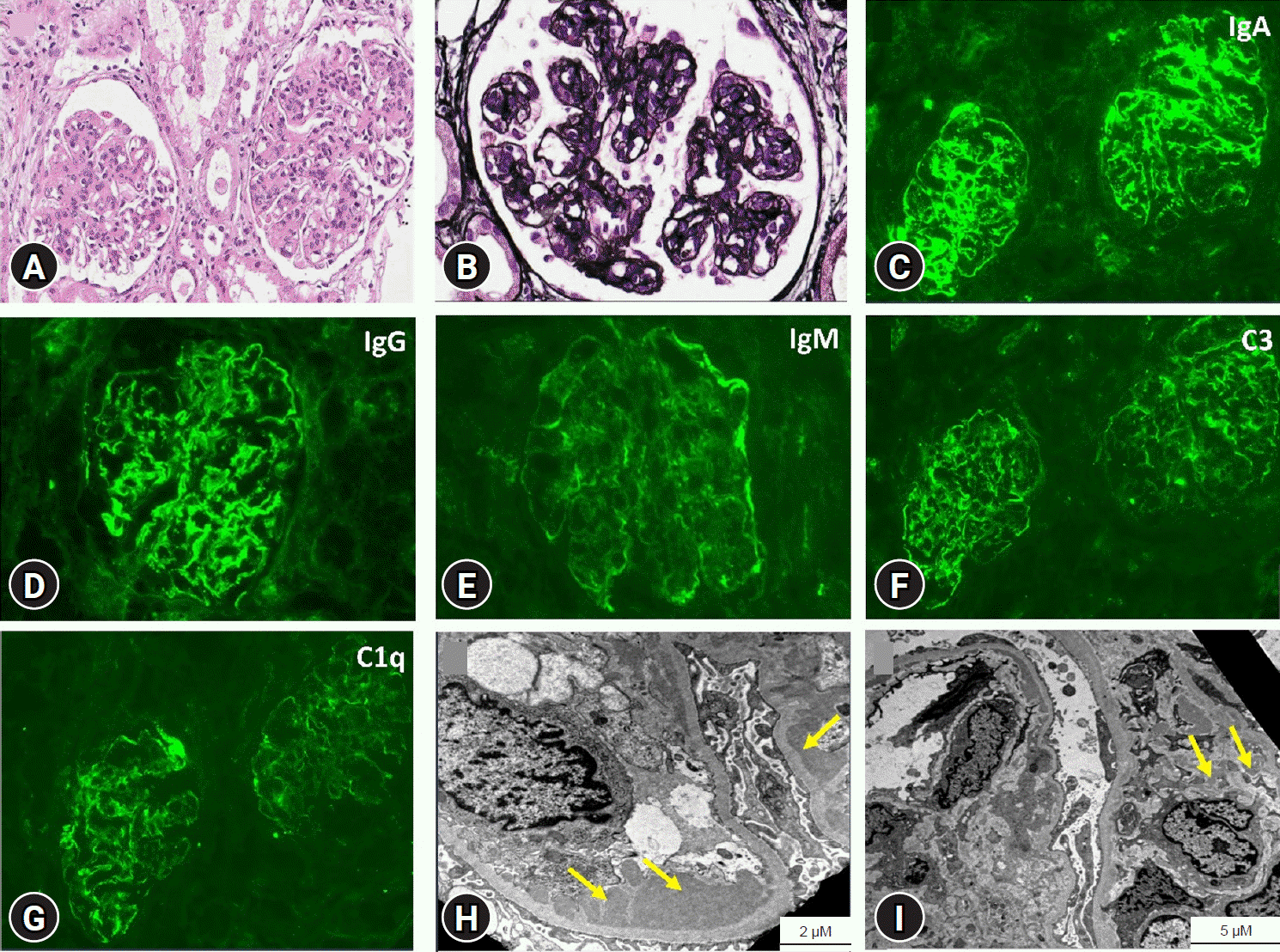 | Fig. 1.Kidney biopsy findings. (A, B) Light microscopy shows diffuse moderate hypercellularity of mesangial and endothelial cells and increased mesangial matrix, which shows a tram-track pattern (A: hematoxylin and eosin stain, ×200; B: methenamine silver Periodic acid-Schiff stain, ×400). (C-G) Immunofluorescence staining shows IgA(3+), IgG(2+), IgM(1+), C3(2+), and C1q(2+) (C, F, G: ×200; D, E: ×400). (H, I) Electron microscopy shows electron-dense deposits (+), moderate mesangial deposits, moderate subendothelial deposits, and marked focal effacement of foot processes. The thickness of the glomerular basement membrane is within the normal limits (H, I). 
|
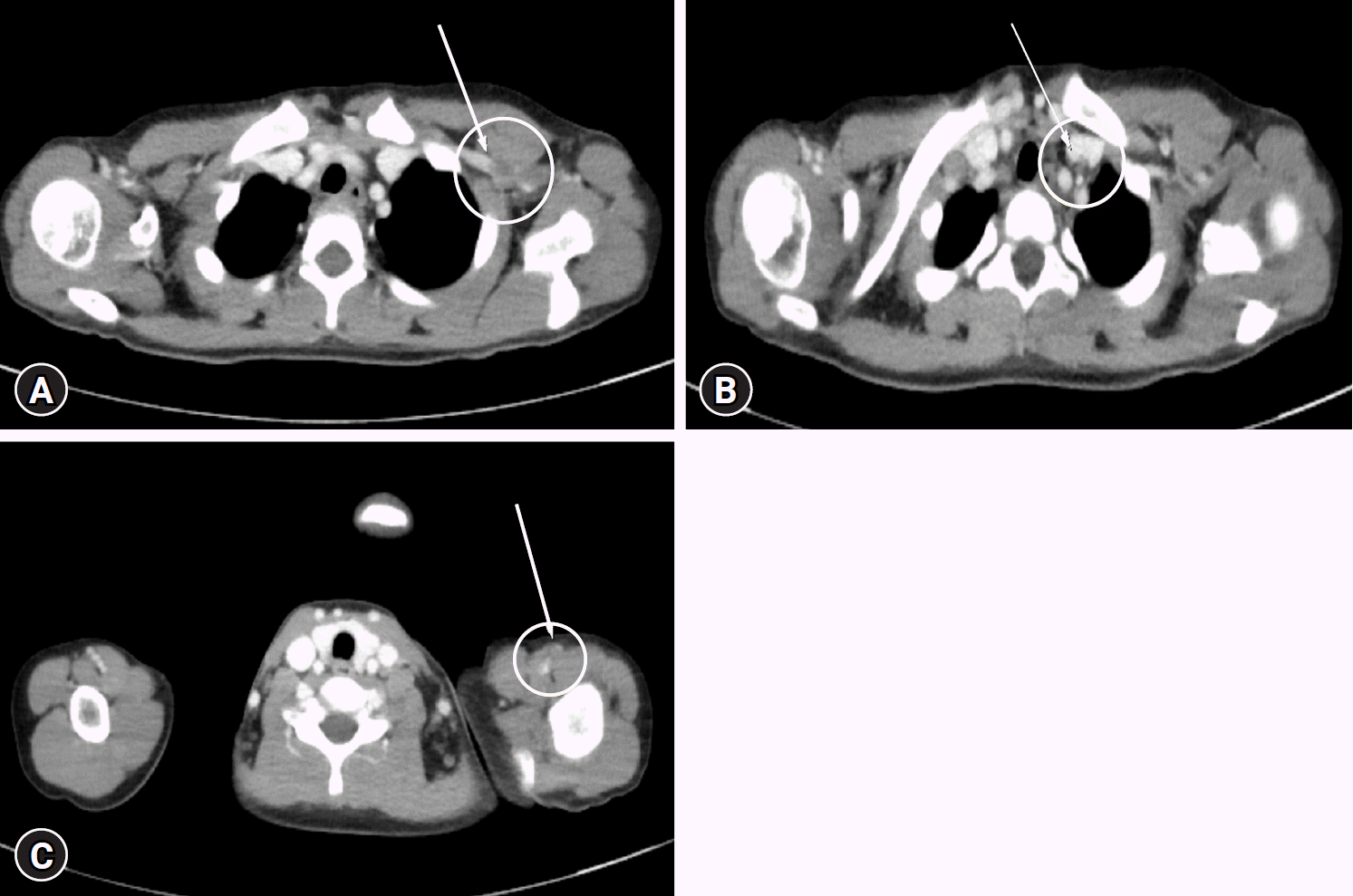 | Fig. 2.Computed tomography angiography with three-dimensional upper extremity artery and vein (with contrast). There are multiple thromboses in the left axillary (A), subclavian (B), and brachial (C) veins (marked by a yellow circle and a white arrow).
|
Table 1.
Laboratory findings of the patient
|
Variable |
Upon IgAN diagnosis |
Upon admission for thrombosis |
Upon LAHPS diagnosis |
Current (post-CPM treatment) |
Reference range |
|
WBC (/μL) |
7,380 |
7,150 |
6,330 |
5,600 |
4,500–14,500 |
|
Hemoglobin (g/dL) |
11.1 |
8.0 |
10.7 |
14.7 |
11.5–15.5 |
|
Platelet (/μL) |
221 K |
226 K |
207 K |
289 K |
130–400 K |
|
BUN/Cr (mg/dL) |
19/0.48 |
32/0.94 |
40/1.4 |
10/0.51 |
- |
|
Cystatin C (mg/L) |
1.17 |
1.69 |
2.29 |
1.26 |
- |
|
eGFR (mL/min/1.73 m2)a)
|
78.9 |
42.2 |
33.6 |
83.0 |
- |
|
Albumin (g/dL) |
2.7 |
2.7 |
3.6 |
1.8 |
3.3–5.2 |
|
Cholesterol (mg/dL) |
409 |
546 |
178 |
Not done |
0–240 |
|
Coagulation panel |
|
|
|
|
|
|
PT INR |
0.89 |
0.90 |
1.56 |
1.16 |
0.8–1.2 |
|
aPTT (sec) |
50.8 |
43.0 |
69.3 |
55.5 |
27.1–37.8 |
|
Fibrinogen (mg/dL) |
325 |
415 |
286 |
425 |
192–411 |
|
Urine laboratory |
|
|
|
|
|
|
UPCR (mg/mg) |
11.60 |
16.55 |
2.30 |
12.24 |
<0.2 |
|
RBC (/HPF) |
≥100 |
≥100 |
≥100 |
10-19 |
0–4 |

Table 2.
Systemic lupus erythematosus-related laboratory profiles
|
Variable |
Upon IgAN diagnosis |
Upon admission for thrombosis |
Upon LAHPS diagnosis |
Current (post-CPM treatment) |
Reference range |
|
C3 (mg/dL) |
82 |
105 |
45 |
125 |
70–150 |
|
C4 (mg/dL) |
10 |
20 |
2 |
19 |
10–35 |
|
ANA |
Positive (homogenous pattern) |
Positive (1:40) |
Positive (>1: 320) |
ND |
Negative |
|
Anti-dsDNA Ab (IU/mL) |
Positive (21.3) |
Negative (<1.0) |
Positive (907.5) |
Negative (<1.0) |
0–7 |
|
Anticardiolipin Ab, IgG (U/mL) |
ND |
Negative |
Positive (71.7) |
Negative |
Negative |
|
Anticardiolipin Ab, IgM (U/mL) |
ND |
Negative |
Positive (24.2) |
Positive (24.7) |
Negative |
|
Anti-β2GPI IgG (U/mL) |
ND |
Negative |
Positive (344.0) |
Positive (33.6) |
Negative (<20) |
|
Anti-β2GPI IgM (U/mL) |
ND |
Negative |
Positive (32.0) |
Positive (30.1) |
Negative (<20) |
|
LA |
Positive |
Positive |
Positive |
Positive |
Negative |
Table 3.
The 2019 European League Against Rheumatism/American College of Rheumatology criteria for SLEa)
|
Domain |
Criteria |
Weight |
Upon IgAN diagnosis |
Upon throm-botic event |
Upon LAHPS diagnosis |
|
Clinical domains |
|
|
|
|
|
Constitutional |
Fever (>38 °C) |
2 |
2 |
0 |
0 |
|
Hematologic |
Leukopenia (<4,000/μL) |
3 |
0 |
0 |
0 |
|
Thrombocytopenia (<100,000/μL) |
4 |
0 |
0 |
0 |
|
Autoimmune hemolysis |
4 |
0 |
0 |
0 |
|
Neuropsychiatric |
Delirium |
2 |
0 |
0 |
0 |
|
Psychosis |
3 |
0 |
0 |
0 |
|
Seizure (generalized or partial/focal) |
5 |
0 |
0 |
0 |
|
Mucocutaneous |
Nonscarring alopecia |
2 |
0 |
0 |
0 |
|
Oral ulcers |
2 |
0 |
0 |
0 |
|
Subacute cutaneous lupus or discoid lupus |
4 |
0 |
0 |
0 |
|
Acute cutaneous lupus |
6 |
0 |
0 |
0 |
|
Serosal |
Pleural effusion or pericardial effusion |
5 |
0 |
0 |
0 |
|
Acute pericarditis |
6 |
0 |
0 |
0 |
|
Musculoskeletal |
Joint involvement |
6 |
0 |
0 |
0 |
|
Renal |
Proteinuria (>0.5 g/24 hr) |
4 |
4 |
4 |
0 |
|
Class II or V LN |
8 |
0 |
0 |
0 |
|
Class III or IV LN |
10 |
0 |
0 |
10 |
|
Immunologic domains |
|
|
|
|
|
Antiphospholipid antibodies |
Anticardiolipin antibodies or anti-β₂GPI or LA |
2 |
2 |
2 |
2 |
|
Complement |
Low C3 or low C4 |
3 |
0 |
0 |
0 |
|
Low C3 and low C4 |
4 |
0 |
0 |
4 |
|
SLE-specific antibodies |
Anti-dsDNA antibody or anti-Smith antibody |
6 |
6 |
0 |
6 |
|
Total score |
|
|
14 |
6 |
22 |


 PDF
PDF Citation
Citation Print
Print



 XML Download
XML Download