

 PDF
PDF Citation
Citation Print
Print



Introduction
The prevalence of hypertension is increasing worldwide. As of 2020, hypertension has been reported to affect 28.4% of Korean adults aged ≥30 years [1]. Although the etiology of hypertension is most commonly primary (essential or idiopathic), in approximately 15% of cases, hypertension develops as a secondary cause [2]. Clinical manifestations suggestive of secondary hypertension include severe or drug-resistant hypertension, new onset hypertension that had been controlled, hypertension diagnosed at a younger age (<30 years) without obesity or a family history of hypertension, and disproportionate target organ damage (e.g., hypertensive retinopathy or left ventricular hypertrophy) [3].
Therefore, efforts to differentiate secondary hypertension in patients with suspected clinical features are crucial. By managing the secondary causes of hypertension, high blood pressure can be normalized to prevent target organ damage. The causes of secondary hypertension include renal diseases (renal vascular or parenchymal diseases), endocrine diseases, and drugs [4]. Hypertension may be the initial clinical manifestation of adrenal, parathyroid, pituitary, or thyroid-dependent metabolic disorders such as primary aldosteronism (PA), pheochromocytoma, Cushing disease (and the resulting Cushing syndrome), hyperdeoxycorticosteronism, hyperparathyroidism, acromegaly, hypothyroidism, hyperthyroidism, and obstructive sleep apnea [2]. This review aims to summarize the clinical signs, evaluation, and treatment of endocrine hypertension, with a focus on adrenal gland disorders.
Primary aldosteronism
PA is the most common cause of endocrine hypertension and is usually diagnosed when the patient is 20 to 60 years of age [5]. The prevalence of PA is approximately 4% to 30% [6,7]; however, less than 2% of cases are detected [8]. The degree of hypertension can be mild to severe and may be resistant to standard pharmacological treatments. Generally, aldosterone levels and blood pressures are higher in patients with aldosterone-producing adenomas than in those with bilateral hyperplasia [5].
1. Clinical manifestations and comorbidities
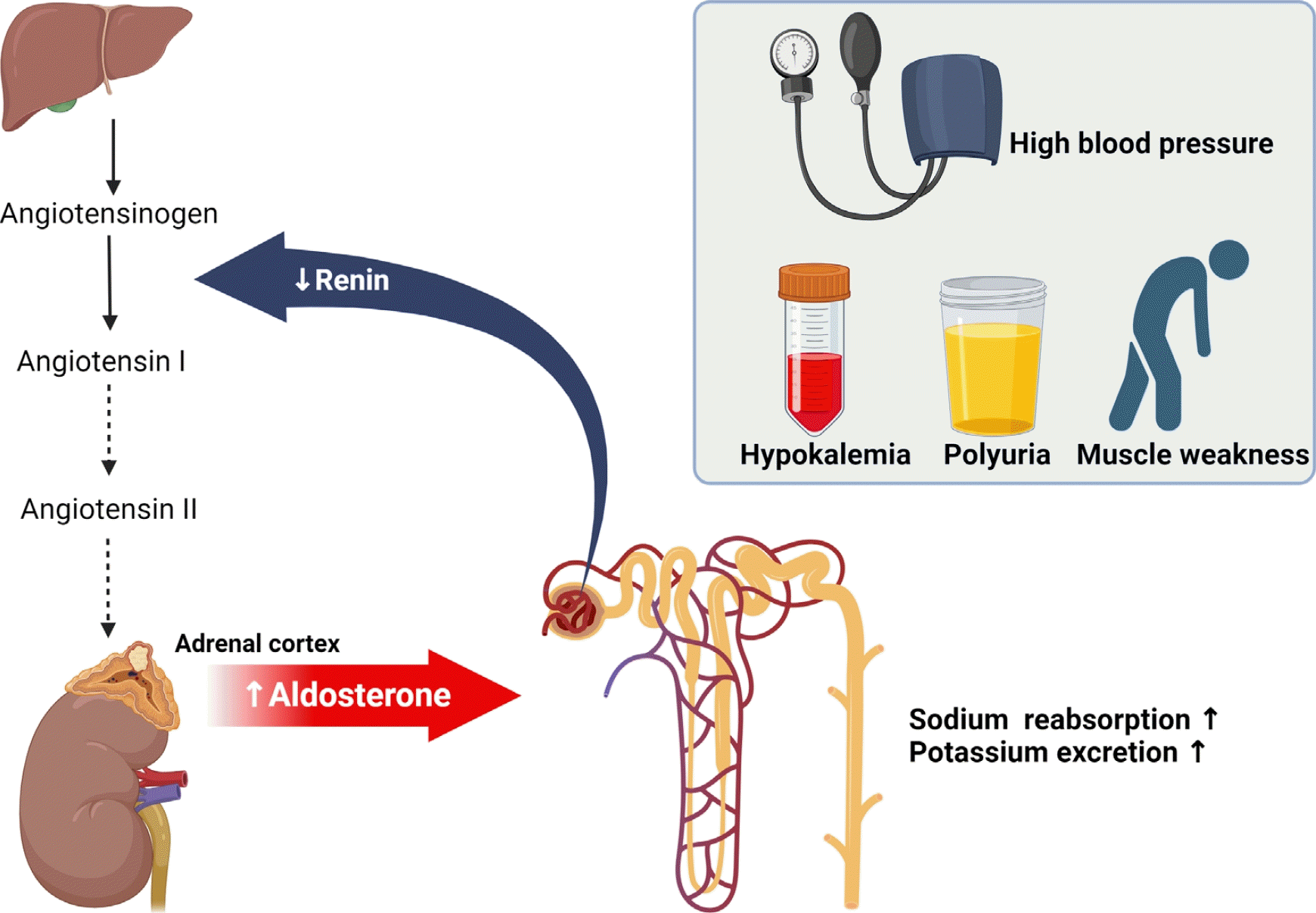
Autonomous aldosterone secretion increases sodium reabsorption in the distal renal tubules and induces water retention, resulting in high blood pressure and renin-angiotensin II suppression [9]. In addition, potassium excretion increases in the distal tubules, causing hypokalemia, which increases renal ammonia production and hydrogen ion excretion, causing metabolic alkalosis [10]. Persistent hypokalemia may decrease the concentrating ability of the kidneys, resulting in polyuria [11]. In other words, PA should be suspected in patients with high blood pressure, headache, hypokalemia, metabolic alkalosis, nephrogenic diabetes insipidus, muscle weakness, or sensory abnormalities (Fig. 1).
Hypokalemia is present in only 28% of patients with PA [12]. Therefore, PA should be considered in the differential diagnosis of all patients with hypertension. The consensus guidelines of the Korean Endocrine Society [13], European Society of Hypertension [14], and American Endocrine Society [15] strongly recommend screening for PA in the cases presented in Table 1. Patients with moderate to severe hypertension that is resistant to standard pharmacological treatments; those with young-onset hypertension (<40 years old); those with hypertension and hypokalemia, adrenal incidentaloma, or atrial fibrillation; and those with a family history of early-onset hypertension, stroke, or PA should undergo screening tests. The primary difference among the three guidelines lies in the blood pressure threshold for PA screening, possibly reflecting the varying thresholds recommended for hypertension diagnosis [16,17]. Further harmonization of these guidelines to establish an optimal blood pressure threshold for PA screening would be advantageous.
PA causes cardiometabolic (obstructive apnea, stroke, myocardial infarction, and atrial fibrillation) [18-20], renal (albuminuria and increased glomerular filtration rate) [21], and bone (increased risk of fracture) complications [22]. According to a meta-analysis comparing the risk of comorbidities, the risk of cardiovascular disease was 1.7 to 3.5 times higher [11] and that of renal complications was 2.0 to 3.4 times higher in patients with PA than in those with essential hypertension [23]. Thus, prompt diagnosis and treatment of PA are important not only to prevent target organ damage but also to improve quality of life.
2. Screening tests
In patients with PA, plasma aldosterone concentration (PAC) and plasma renin activity (PRA) decrease. Compared to PAC and PRA, the aldosterone-to-renin ratio (ARR) has higher sensitivity and specificity as a screening test for PA [2]. An ARR >30 and a PAC >15 ng/dL indicate a clear increase in aldosterone autonomic secretion [10,13]. Some antihypertensive drugs might induce false-positive (beta-blockers) or false-negative (angiotensin receptor blockers, angiotensin-converting enzyme inhibitors, and mineralocorticoid receptor antagonists [MRAs]) results; therefore, it is recommended to measure the ARR 2 to 6 weeks after withdrawing antihypertensive medications (especially MRAs [spironolactone and eplerenone]). The antihypertensive drugs can be switched to non-dihydropyridine calcium channel blockers, vasodilators, or alpha-blockers that have minimal effects on ARR [14,15]. Case detection testing can be completed without the discontinuation of antihypertensive medications [24]. For example, in cases where PRA is suppressed despite MRA treatment, the MRA can be administered at a subtherapeutic dose, and the patient can undergo screening and confirmatory tests without drug discontinuation.
3. Confirmatory tests
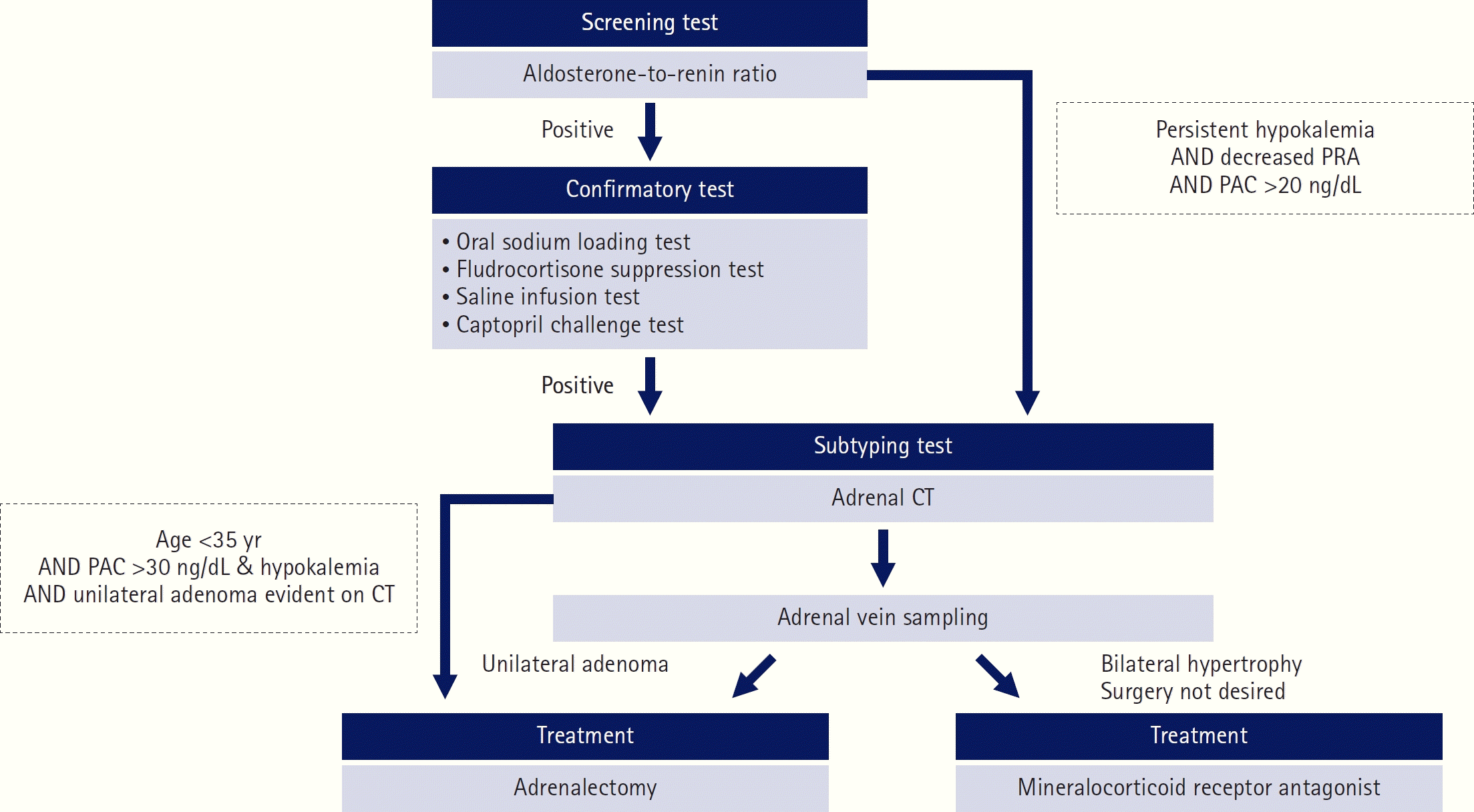
Tests are required to confirm that aldosterone production is not inhibited by acute volume expansion following the suppression of angiotensin II synthesis. Four confirmatory tests are recommended, including the oral sodium loading test, fludrocortisone suppression test, saline infusion test, and captopril challenge test; non-suppression of 24-hour urine aldosterone or PAC is diagnostic [13,14]. The saline infusion test is an accurate and cost-effective confirmatory test performed with patients in the seated position (superior to the supine position with higher sensitivity) [25,26]. An immunoassay is a time- and cost-effective method for measuring PAC; however, it has a lower specificity for diagnosing PA than liquid chromatography-mass spectrometry [27]. Subtype testing (adrenal computed tomography [CT] and adrenal venous sampling [AVS]) is performed without a confirmatory test in cases of persistent hypokalemia, decreased PRA, or PAC >20 ng/dL [15]. The diagnostic algorithm for PA is shown in Fig. 2.
AVS is the gold standard for categorizing and distinguishing unilateral and bilateral aldosteronism. The selectivity index, lateralization index, contralateral ratio, and ipsilateral ratio are calculated by measuring the aldosterone and cortisol levels in the bilateral adrenal veins and inferior vena cava [15]. The success rate is higher in the left adrenal vein than in the right adrenal vein owing to anatomical differences. In cases of unilaterally successful AVS, measuring the relative aldosterone secretion index may be useful [28]. Adrenalectomy may be performed immediately without AVS in patients aged <35 years with marked hyperaldosteronism (PAC >30 ng/dL), remarkable hypokalemia, and unilateral adrenal tumors evident on adrenal CT [13,15].
Approximately 5% of patients with PA have genetic mutations associated with familial hyperaldosteronism (FH) [29]. FH gene mutation testing is recommended (e.g., chimeric CYP11B1/B2, CLCN2, KCNJ5, and CACNA1H) in patients aged <20 years diagnosed with PA or stroke and/or with a family history of PA [14,15]. Nearly all patients with FH have bilateral adrenal disease that cannot be cured with unilateral adrenalectomy.
4. Current and future perspectives for treating primary aldosteronism
Treatment aims to normalize blood pressure and correct hypokalemia to reduce cardiovascular complications and mortality. Adrenalectomy is the first-line treatment for unilateral adrenal adenomas. In a single-center cohort study of 93 patients who were surgically treated, the 1-year and 9-year biochemical remission rates were 100% and 77.4%, respectively. Nonclassical histology demonstrated a higher long-term recurrence rate than classical histology [30]. Postoperatively, blood pressure and hypokalemia improved in approximately 100% of the patients [31]. In addition, adrenalectomy induced regression of left ventricular hypertrophy [32] and reversed albuminuria [33]. Younger female patients have been reported to have a higher chance of being cured; however, older men and women may also benefit clinically [34]. However, outcomes should be assessed at 3 months and at 6 to 12 months postoperatively, and reassessments should be performed annually [34].
MRA therapy is recommended for bilateral adrenal hypertrophy or when surgery is not desired [35]. Spironolactone is the first-line MRA therapy in Korea [13]. Recently, a nonsteroidal MRA (finerenone) has been recognized as more tolerable than steroidal MRAs (spironolactone and eplerenone) [36]. Steroidal MRAs are widely distributed in the kidney and may induce hyperkalemia and worsen kidney function [37]. In addition, their affinity for androgen receptors results in gynecomastia, breast pain, and erectile dysfunction [38,39]. In contrast, finerenone is distributed equally in the kidneys and heart [40] and has no affinity for androgen receptors, attenuating electrolyte imbalance and sexual side effects [41]. However, there is limited clinical evidence supporting the use of nonsteroidal MRAs for the treatment of PA. Phase IV trials on the blood pressure-lowering effect and safety of finerenone in patients with PA have been completed (NCT05924620) [42] and are ongoing (NCT06457074) [43], and their results are expected. In addition, whether finerenone can protect against end-organ damage remains unclear, and further studies are needed [36].
Pheochromocytoma and paraganglioma
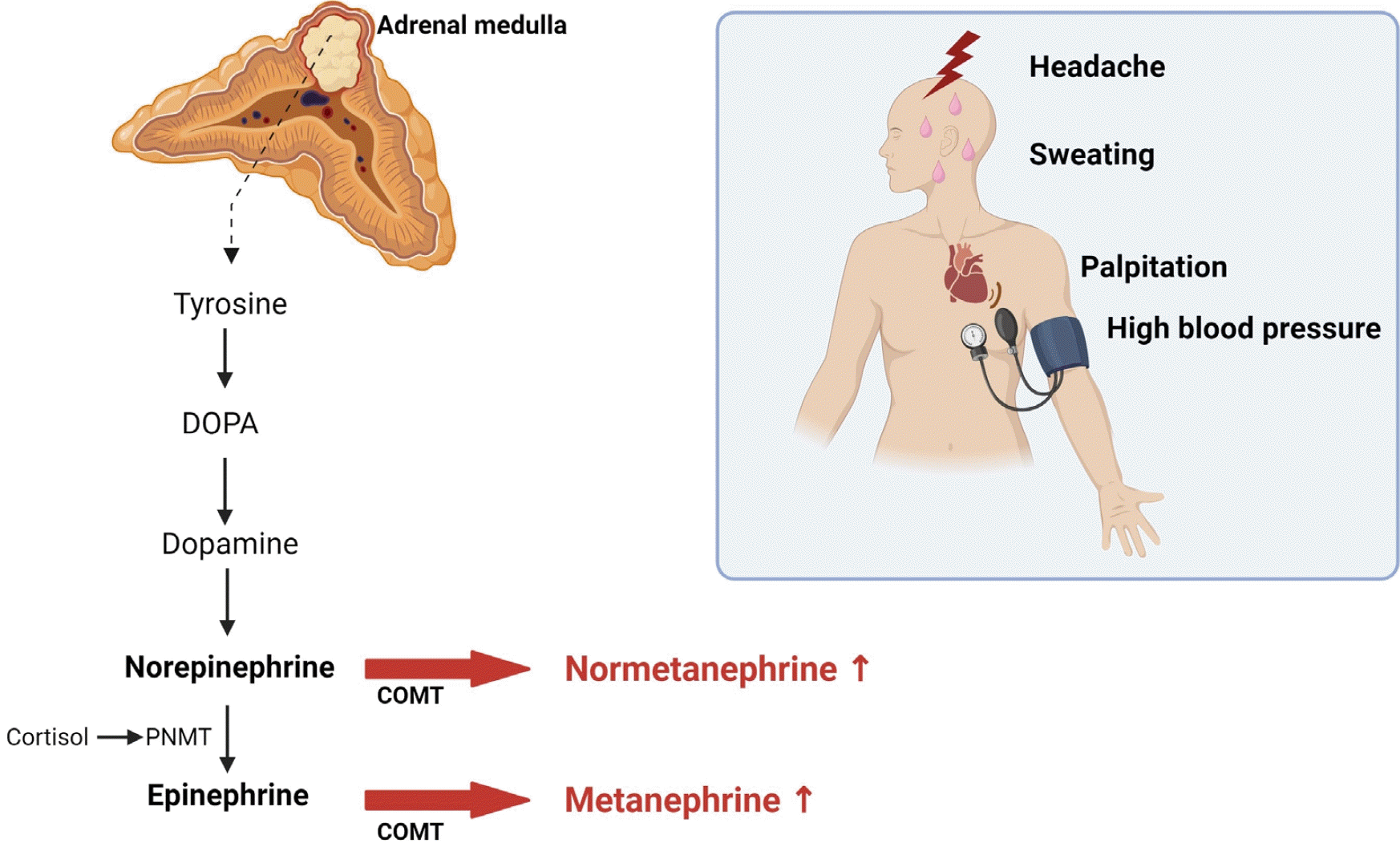
Pheochromocytoma and paraganglioma (PPGL) are rare causes that account for 0.01% to 0.2% of cases of hypertension [44], occurring equally in men and women, primarily between 30 years and 50 years of age [45]. In PPGL, catecholamines (dopamine, norepinephrine, and epinephrine) are secreted by adrenal medullary chromaffin cells (80%–85%) and extra-adrenal chromaffin cells (15%–20%) [46]. Catechol-O-methyltransferase exists in the adrenal medulla and metabolizes catecholamines to normetanephrine and metanephrine. Under normal conditions, only small amounts of catecholamines are metabolized by this pathway; however, when PPGL occur, it becomes the main metabolic pathway [47] (Fig. 3).
1. Clinical manifestations
The classic triad of PPGL includes paroxysmal headaches, palpitations, and sweating [48]. Persistent or paroxysmal hypertension is present in 80% of patients [49]. However, PPGL often present with atypical symptoms, such as myocardial infarction with normal coronary arteries or systemic inflammatory response syndrome [50,51]. In the 2000s, more cases without typical symptoms were diagnosed by incidentally discovering an adrenal mass on abdominal CT [52].
PPGL are curable when correctly diagnosed and treated but can be fatal if undiagnosed or inadequately treated. Routine screening is not recommended for all patients with hypertension; however, screening should be performed once there are clinical signs of PPGL (Table 2), regardless of blood pressure [46,53].
2. Detection tests
An initial measurement of plasma-free or urine-fractionated metanephrines (metanephrine and normetanephrine) is recommended when pheochromocytoma is clinically suspected [46] because, unlike catecholamines secreted by tumors, their metabolites are continuously released [54]. Liquid chromatography-mass spectrometry and electrochemical detection methods are more accurate than immunoassay methods [55,56]. When measuring plasma-free metanephrines, it is recommended to draw blood after the patient rests in the supine position for 30 minutes [46]. Acetaminophen and tricyclic antidepressants (TCAs) are representative drugs that cause false-positive results by directly interfering with liquid chromatography assays and inhibiting norepinephrine reuptake, respectively [57]. TCAs and antipsychotic drugs should be gradually reduced and discontinued for 2 to 4 weeks before testing. If the test is performed without terminating the medication and the results are normal, further evaluation is unnecessary. Moreover, physiological stress (e.g., intensive care settings) can markedly increase plasma or urine metanephrines [58]. The test accuracy is comparable between plasma and urine tests, and the sensitivity is ≥95%; thus, PPGL can be excluded with high confidence if normal values are obtained [59]. The specificities of plasma and urine tests are reported to be 85% and 98%, respectively. Therefore, false-positive results may be reduced if 24-hour urinary metanephrine levels are measured [60].
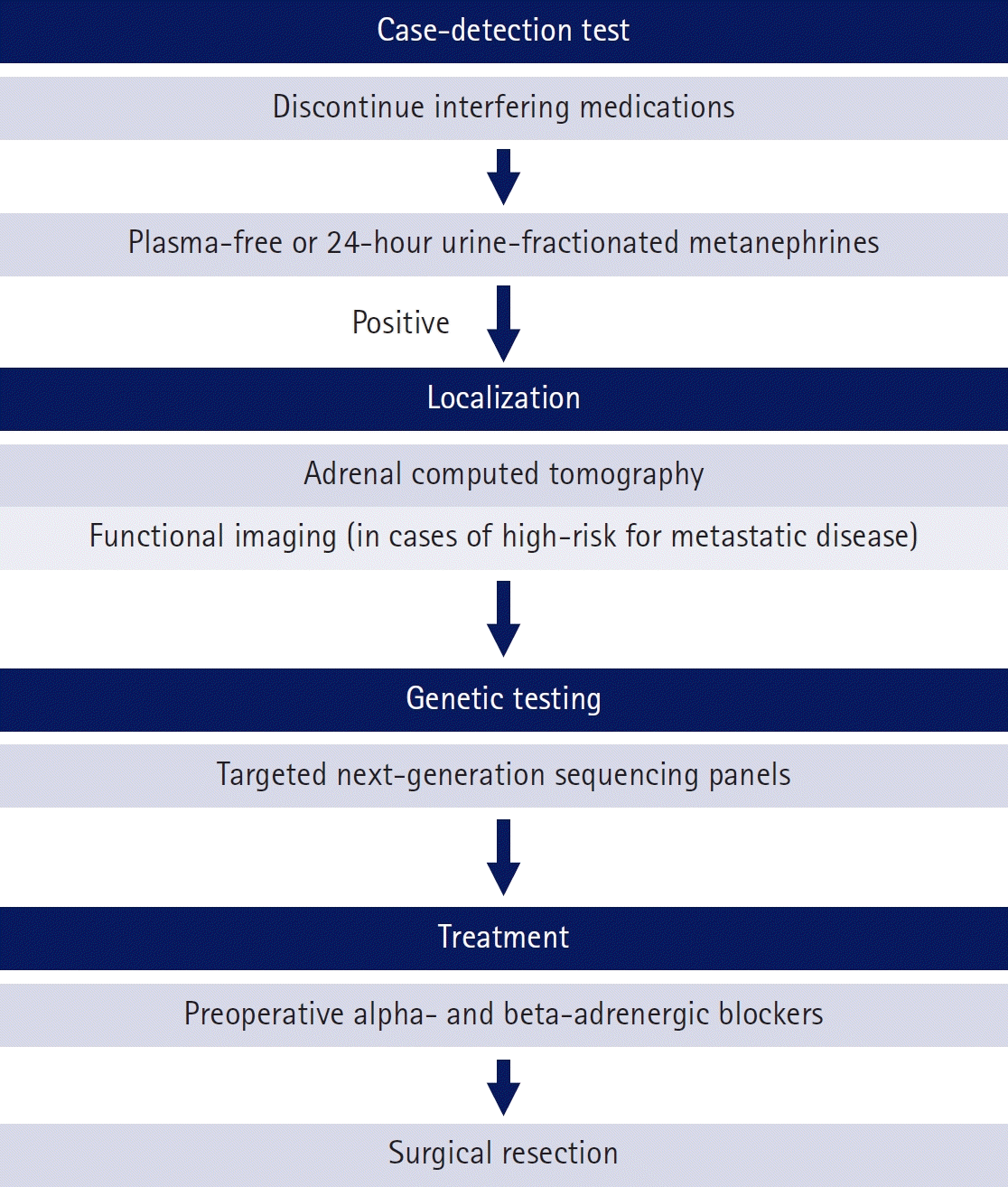
Imaging tests, preferably CT, are performed to locate the tumor. Eighty-five percent of PPGL are located in the adrenal glands, and 95% of extra-adrenal tumors are located in the abdomen and pelvis [53,61]. When CT attenuation is ≤10 Hounsfield unit, it indicates the presence of a lipid-rich mass and PPGL are excluded; hence, biochemical testing is unnecessary [62]. In cases of larger tumors (>5 cm, extra-adrenal, bilateral, multifocal, and recurrent disease), the possibility of metastasis should be considered, and functional imaging is recommended, including 68Ga-DOTA-somatostatin analog positron emission tomography (PET)/CT, 18F-fluoro-ʟ-DOPA PET/CT, 18F-fluorodeoxyglucose PET/CT, and 123I/131I-metaiodobenzylguanidine (MIBG) scintigraphy/single-photon emission CT [63]. The diagnostic algorithm for PPGL is shown in Fig. 4.
3. Genetic testing
PPGL are the most common hereditary tumors in humans and 30% of patients with PPGL harbor germline mutations [46]. Therefore, all patients diagnosed with PPGL should undergo genetic testing regardless of family history [64]. Related genetic diseases include multiple endocrine neoplasia type 2, von Hippel-Lindau (VHL) syndrome, neurofibromatosis (NF) type 1, and hereditary paraganglioma syndrome. With the advent of next-generation sequencing (NGS) panels, all pheochromocytoma-related genes can be screened using a single test. The 10 basic NGS panels recommended in Korea are fumarate hydratase, myc-associated protein X, NF type 1, rearranged during transfection (RET), succinate dehydrogenase (SDH)A, SDHB, SDHC, SDHD, transmembrane protein 127, and VHL [63]. For a person who is asymptomatic and at risk of disease based on a family history of PPGL, surveillance should be started 5 years earlier than the age of family diagnosis [65].
4. Current and future perspectives for treating pheochromocytoma and paraganglioma
The treatment goal for PPGL is complete tumor resection, which requires a multidisciplinary team of experienced internists, anesthesiologists, and surgeons to ensure prompt and safe surgical treatment. Preoperative medical treatment is essential for controlling hypertension and tachycardia and restoring circulating volume [66]. Administration of alpha-adrenergic blockers, preferably phenoxybenzamine or doxazosin, 7 to 14 days preoperatively, is considered the first choice. A beta-adrenergic blocker can be added, typically 3 days preoperatively, if tachycardia persists after the initiation of an alpha-adrenergic blocker [67]. Clinicians should be aware that beta-adrenergic blockers must be administered after alpha-adrenergic blockers to prevent hypertensive crisis [68]. Alternatives to alpha- and beta-adrenergic blockers include calcium channel blockers and metyrapone, which achieve normotension and inhibit catecholamine synthesis, respectively [67]. A minimally invasive approach, such as laparoscopic and robot-assisted adrenalectomy, can be safely performed for localized adrenal pheochromocytoma [69].
Combination systemic chemotherapy (cyclophosphamide, vincristine, and dacarbazine) has emerged as a standard treatment option for metastatic progressive PPGL [70]. Recently, 131I-MIBG (Azedra, Progenics Pharmaceuticals, Inc.) received approval for patients >12 years old with iobenguane scan-positive advanced unresectable PPGL [71]. There is evidence that 131I-MIBG lowers blood pressure and shrinks tumors [72], and the imaging response at 3 to 6 months predicts improved survival for 4 years [73]. A phase II clinical trial indicated that patients with germline variants in the subunits of SDH or RET may benefit from 37.5 mg of sunitinib [74,75]. The optimal treatment for metastatic PPGL can be determined based on the accumulation of additional data.
The European guideline recommends assessing the levels of metanephrines and 3-methoxytyramine (±chromogranin A) in plasma or urine 2 to 6 weeks postoperatively [76]. In cases in which these levels are elevated, an imaging test is recommended 3 months postoperatively to assess complete resection. Surgical resection of PPGL cannot guarantee a complete cure. Therefore, all patients who undergo surgery for PPGL should be followed up for 10 years to monitor recurrence. Patients at high risk (young age, gene mutation, or larger tumors) require lifelong follow-up.
Conclusion
PA and PPGL have characteristic clinical symptoms and can be treated surgically or managed with specific medications. Screening for PA, which is the most common cause of endocrine hypertension, should be performed at least once in all patients with suspected secondary hypertension. Delays in PPGL diagnosis tend to have fatal consequences. Therefore, biochemical testing must be performed even with the slightest suspicion, regardless of blood pressure. A multidisciplinary approach is essential for managing PA and PPGL.
 XML Download
XML Download