

 PDF
PDF Citation
Citation Print
Print



Introduction
Adrenal cortical carcinoma (ACC) is an uncommon tumor that originates in the adrenal cortex and accounts for ≤10% of adrenal incidentalomas [1-5]. Its incidence is approximately one case to two cases per million individuals annually, with biochemical evidence of adrenocortical hormone production in approximately 45% to 70% of cases. Most patients presenting with hormone excess commonly exhibit hypercortisolism as their primary symptom, which is observed in as many as 50% to 80% of hormone-secreting ACCs, with 24% exhibiting concurrent virilization [6,7]. A few of these tumors primarily release aldosterone, with only 2.5% of all ACCs exclusively producing aldosterone [8]. Pheochromocytomas are catecholamine-secreting paragangliomas that develop in adrenal or extra-adrenal locations [9]. Cases of pheochromocytoma concurrent with ACC have been documented, yet it is uncommon for ACCs to manifest clinically as pheochromocytomas [10]. This case underscores an uncommon occurrence of ACC that clinically and biochemically mimics pheochromocytoma with cosecretion of aldosterone and cortisol.
Case
Ethics statement: This study was reviewed by the Institutional Review Board (IRB) of Konyang University Hospital (IRB No: 2024-05-002-003). Written informed consent was obtained from the patient to participate in the study. Informed consent for publication of the patient’s clinical details and images was also obtained from the patient.
A 53-year-old man presented to the Emergency Department with a referral from another hospital for embolization of a ruptured right adrenal mass with retroperitoneal hematoma. The patient had been evaluated for a sudden-onset sharp, stabbing right abdomen pain that started 5 hours earlier after lifting a heavy object. His medical history was significant for hypertension diagnosed 1 year earlier, which was not treated, and a 5 cm-sized adrenal adenoma diagnosed 7 years earlier. Laboratory tests conducted 7 years ago to evaluate the adrenal incidentaloma indicated potential primary hyperaldosteronism with aldosterone levels at 20.2 ng/dL, which were at the high end of normal (range, 6.7–33.5 ng/dL), and suppressed plasma renin activity of 0.82 ng/mL/hr (range, 1–30.0 ng/mL/hr). Further testing 7 years ago confirmed excess cortisol production independent of adrenocorticotropic hormone (ACTH), with an ACTH level of 2.17 pg/mL (range, 7.2–63.3 pg/mL), and a 1-mg overnight dexamethasone suppression test revealed a cortisol level of 11.63 μg/dL (range, <1.8 μg/dL) (Table 1). The last follow-up appointment at another hospital 2 years earlier did not reveal any change in tumor size or hormone work-up. There was no family history of genetic syndromes that predisposed the patient to ACC. The patient had an elevated blood pressure of 130/100 mmHg, consistent with untreated hypertension.
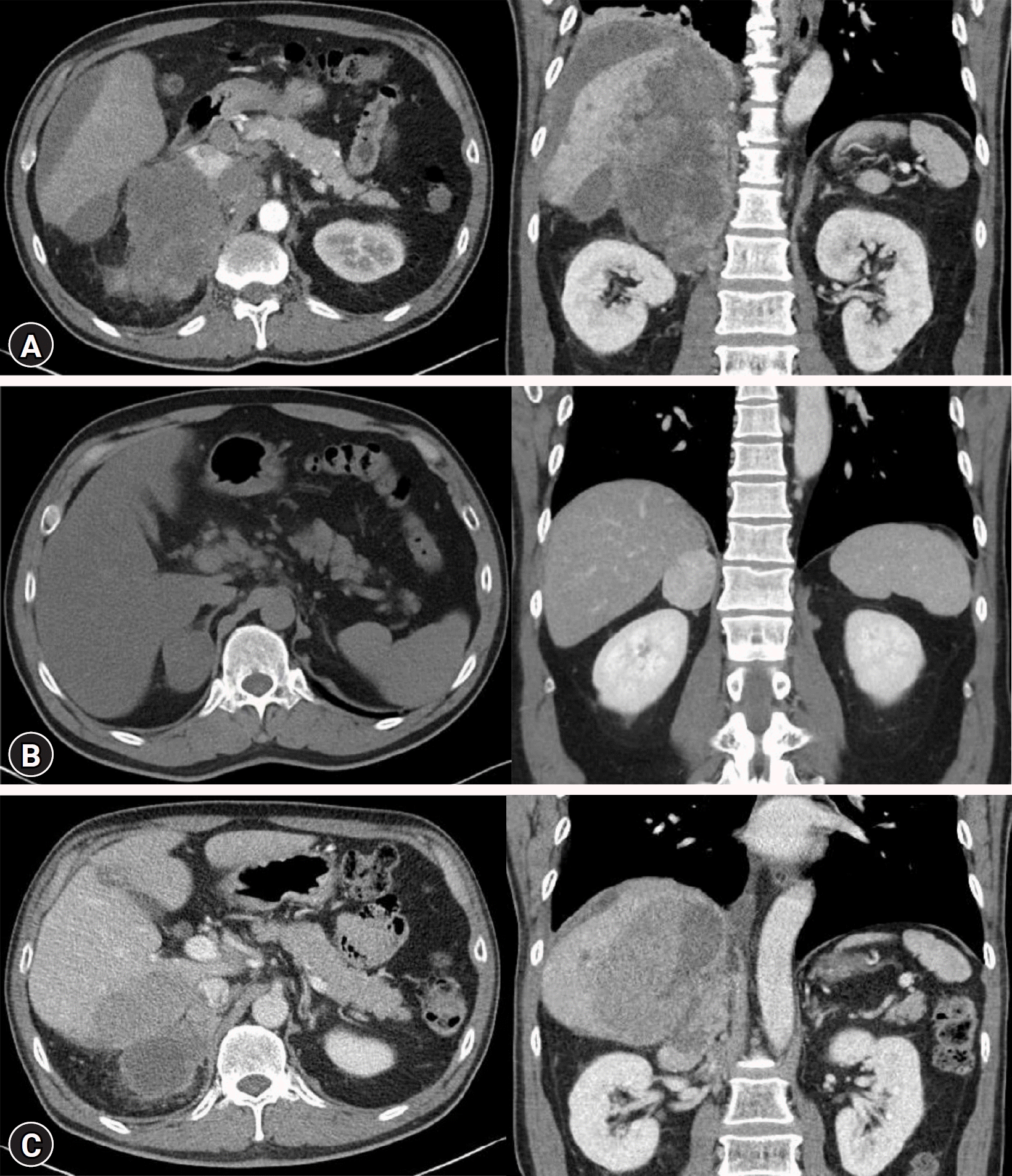
Other vital signs were normal. Physical examination revealed pale conjunctiva and tenderness in the right upper quadrant and epigastrium. No signs of hypertensive urgency, excess cortisol levels, or bruising were observed. No abdominal striae, central obesity, or abnormal fat distribution was observed. Intravenous access was established through the right jugular vein, and a basic metabolic panel indicated normal creatinine levels. However, mild hypokalemia was observed with a potassium level of 3.46 mmol/L (3.0 mEq/L; range, 3.5–5.1 mmol/L), along with elevated liver enzymes at 166/108 IU/L (40/41 IU/L). Abdominal computed tomography (CT) revealed a heterogeneous 19×11×15 cm right adrenal mass invading the right lobe of the liver, inferior vena cava, retrocaval lymph nodes, and aortocaval lymph nodes (Fig. 1). The left adrenal gland was normal. The patient was referred to the Radiology Department for angioembolization, which was performed without complications. Antihypertensive medication (intravenous nicardipine) was initiated because of an elevated blood pressure of 160/120 mmHg.
Because of the presence of hypertension and hypokalemia, serum aldosterone levels and plasma renin activity were evaluated, which revealed normal aldosterone at 11.2 ng/dL (range, 6.7–33.5 ng/dL) and non-suppressed plasma renin activity of 7.96 ng/mL/hr (range, 1–30.0 ng/mL/hr). A saline infusion test suggested primary hyperaldosteronism with a post-infusion aldosterone level of 8.5 ng/dL (range, <6 ng/dL seated). Additional laboratory testing confirmed excessive cortisol production unrelated to ACTH, with an ACTH level of 2.17 pg/mL (range, 7.2–63.3 pg/mL). Furthermore, results from a 1-mg overnight dexamethasone suppression test indicated a cortisol level of 18.3 μg/dL (range, <1.8 μg/dL). Plasma normetanephrine levels were elevated to 2.98 nmol/L (range, <0.64 nmol/L). No further hormonal testing was performed.
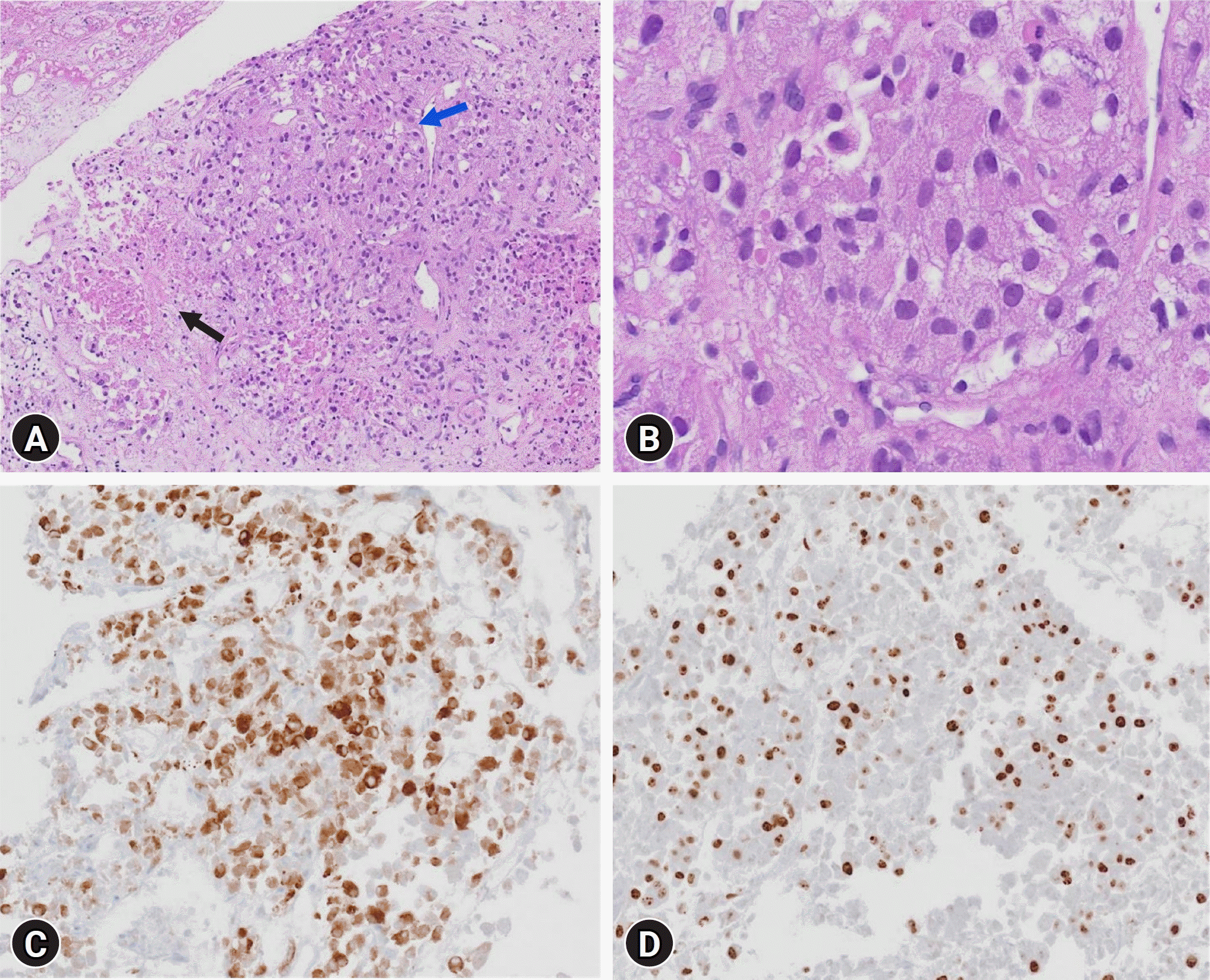
Due to the presence of triple hormone hypersecretion and imaging findings suggestive of malignancy, adrenalectomy was considered. However, given the severity of the local invasion, which would make clear margin resection difficult, and considering the low risk of rupture or rebleeding after embolization of all adrenal arterial inflows, palliative surgical resection was not performed. A biopsy of the right adrenal mass was performed for pathological confirmation to initiate palliative chemotherapy. The results showed tumor necrosis and positivity for inhibin α and vimentin, which was consistent with the diagnosed condition. Ki-67 staining revealed markedly increased proliferation (59.9% positive in 50 high-power fields) (Fig. 2). Next-generation sequencing (CancerSCAN [solid tumor L2]) revealed mutations in TP53 and CTNNB1. Therefore, the patient was diagnosed with stage T4N1M1 ACC, and the first cycle of adjuvant mitotane therapy was started, which was titrated to 500 mg four times per day. Adjuvant treatment with doxorubicin, cisplatin, and etoposide was also initiated because of the aggressive nature and rapid progression of the high-grade disease. The patient's blood pressure remained within normal range and the hypokalemia resolved without subsequent hyperkalemia or decline in kidney function. The patient recovered smoothly and experienced an uncomplicated clinical course after embolization. The patient was discharged on antihypertensive therapy, mitotane, and codeine for pain control. His 3-month follow-up CT scan showed decreased sizes of the right adrenal gland, right para-aortic lymph nodes, and metastatic nodules in the right lobe of the liver. The patient is still alive (after 4 months). This case emphasizes the importance of maintaining a high level of suspicion of ACC in adrenal incidentalomas that mimic pheochromocytomas or present with elevated levels of multiple cortical hormones (aldosterone and cortisol).
Discussion
ACC is an infrequent yet markedly aggressive cancer, occurring at a rate of 0.7 to 2.0 cases per million people annually [1]. Approximately 60% of these tumors are functional, with hypercortisolism being the most prevalent, affecting approximately half of all functional cases [11,12]. The remaining subtypes consist of mixed hormone-secreting tumors (25%), tumors secreting isolated sex hormones (20%), and tumors secreting isolated aldosterone (7.9%) [13]. According to a cohort study, patients with functional tumors experienced poorer long-term survival outcomes [13]. Currently, there are no available data indicating whether multiple hormone secretion affects the likelihood of malignancy [14].
Current guidelines advise screening all adrenal masses for biochemical excess, particularly hyperaldosteronism in cases of hypertension and/or hypokalemia, hypercortisolism, or catecholamine excess [4]. Additionally, patients with an abnormal aldosterone-to-renin ratio should undergo further confirmatory testing, which may include a saline challenge, oral salt loading, captopril challenge, or fludrocortisone suppression test. If spontaneous hypokalemia is present along with undetectable renin and a plasma aldosterone concentration >20 ng/dL (550 pmol/L), further confirmatory testing may be unnecessary [15]. In the current case, confirmatory testing was performed because of hypertension and hypokalemia; however, adrenal vein sampling was not possible due to rupture. In addition to aldosterone, it is essential to measure plasma or urinary metanephrines and conduct a 1-mg overnight dexamethasone suppression test (using a serum cortisol cutoff of ≤1.8 µg/dL [≤50 nmol/L]) in all patients with an adrenal mass since as many as 50% of patients with adrenal incidentalomas show laboratory evidence of mild autonomous cortisol secretion (MACS) [4,16]. In the present case, the serum cortisol level after the overnight dexamethasone suppression test indicated MACS, with a value of 18.3 µg/dL, which is ten times the serum cortisol cutoff of ≤1.8 µg/dL.
This case revealed a functional tumor with hypercortisolism, hyperaldosteronism, and elevated normetanephrine levels. There are only three reported instances of ACC producing metanephrine and five cases of dual hormone-secreting ACC with a combination of aldosterone and steroid hormones documented in the literature [14,17]. However, to the best of our knowledge, this is the first reported case of triple hormone-secreting ruptured ACC with hypercortisolism, hyperaldosteronism, and elevated normetanephrine levels. Six cases of ACC with dual secretion of aldosterone and cortisol have been reported. The proposed mechanism is believed to arise from genetic overlap between the genes responsible for coding 11β-hydroxylase and aldosterone synthase. This overlap may lead to the formation of a hybrid gene, resulting in heightened secretion of both enzymes, and consequently, both hormones [14].
Regarding metanephrine-producing ACC, only four cases have been documented in the literature. In one of these cases, an elevated urine metanephrine level was observed, although it was not double the normal value [17-19]. In all four documented cases, metanephrine-producing ACC displayed histological characteristics typical of ACC, as observed in the present case. However, none of these patients had concomitant hyperaldosteronism. Three reported cases of mixed corticomedullary carcinoma involved solitary neoplasms comprising adrenocortical and chromaffin cells. Preoperatively, laboratory tests in one case confirmed both cortical and medullary hypersecretion, whereas only cortical hypersecretion was confirmed in the other two cases [20]. Consequently, it is evident that clinical and biochemical findings do not consistently match histological findings.
This case report highlights the importance of thorough clinical and biochemical hormone assessments in patients with adrenal masses because ACC can manifest with various hormone elevations.
 XML Download
XML Download