

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Thyroid hormones (TH), including triiodothyronine and thyroxine, are key regulators of liver metabolism in mammals [1]. These substances are critical for promoting hepatic lipid catabolism, which leads to increased β-oxidation and ketogenesis in both rodents and humans [1]. Consequently, low levels of TH in humans are linked to hepatic steatosis, signaling the onset of metabolic dysfunction-associated steatotic liver disease (MASLD) [2-4]. MASLD, formerly known as non-alcoholic fatty liver disease, encompasses a range of lipid metabolic disorders in the liver. This spectrum progresses from benign steatosis to metabolic dysfunction-associated steatohepatitis (MASH), which is characterized by liver inflammation and elevated risk of developing diabetes, cirrhosis, and hepatocellular carcinoma (HCC) [5-7]. Currently, pharmacological treatments for MASLD are limited [8]. While it was previously believed that TH primarily induced the expression of hepatic genes involved in β-oxidation and mitochondrial biogenesis, research from the past decade has shown that TH-induced macroautophagy, hereafter referred to as autophagy, is also crucial in regulating TH-induced lipid catabolism and mitochondrial energetics in the liver [9]. Autophagy is a cellular recycling process that involves the lysosome-mediated degradation of intracellular macromolecules and organelles [10]. Although autophagy is generally non-selective, certain stimuli can trigger highly selective forms that are essential for maintaining cellular homeostasis [11]. TH-induced autophagy represents one such selective process, capable of specifically targeting lipid droplets (LDs; lipophagy) [12], mitochondria (mitophagy) [13], and protein aggregates (aggrephagy) [14]. With the recent U.S. Food and Drug Administration (FDA) approval of the TH analogue Resmetirom [15] for the treatment of MASLD/MASH and given the clinical evidence of impaired hepatic autophagy in MASLD [16-18], understanding the observed beneficial role of TH-induced selective autophagy on MASLD is imperative. Furthermore, the molecular mechanisms involved must be delineated.
Go to : 
MECHANISM OF AUTOPHAGY INDUCTION BY THYROID HORMONE IN THE LIVER
The induction of autophagy in response to acute stimuli, such as pathogens or sudden energy depletion, involves rapid changes in the activity of cellular nutrient- and energy-sensing kinases. These kinases, specifically mechanistic target of rapamycin complex 1 (mTORC1) and AMP-activated protein kinase (AMPK), initiate the formation of autophagosomes within cells [19]. This acute induction typically occurs within minutes and does not require new mRNA or protein synthesis. The process includes the formation of autophagosomes, the engulfment of cellular cargo, and the subsequent fusion of the autophagosomes with lysosomes. This sequence of events is facilitated by a family of molecules known as autophagy-related proteins (ATGs) [20], which are present in cells in sufficient quantities to initiate autophagy. However, with chronic stimulation, autophagy can rapidly deplete these protein levels, necessitating the synthesis of new ATGs. This synthesis requires the activation of nuclear transcription [21]. Chronic induction of autophagy is often mediated by various nuclear hormone receptors and transcription factors [21].
TH induces intracellular metabolic changes by binding to TH receptors (THRs), a class of nuclear hormone receptors [1]. The two primary types of THRs, THRα and THRβ rapidly initiate the transcription of target genes upon TH binding [1]. In vitro studies in hepatic cells and in vivo studies in animal models have shown that THRs are essential for TH-induced autophagy in hepatocytes [12]. Moreover, mice with a mutant THR that cannot bind TH exhibit reduced basal liver autophagy under euthyroid conditions, comparable to the levels observed in hypothyroid wild-type mice [12]. The suppression of basal autophagy in the absence of TH, as well as in the presence of a mutant THR, is thought to involve THR interaction with a nuclear corepressor, nuclear receptor corepressor 1 (NCOR1), which forms a complex with histone deacetylase 3 (HDAC3) [12]. Although THRs are necessary to stimulate TH-induced autophagy, no reports have indicated direct binding of THRs to the promoters of autophagy-related genes. Instead, the binding of TH to THRs likely induces the expression of other transcription factors, which then bind to the promoters of autophagy genes. Certain nuclear receptors and transcription factors that may participate in regulating TH-induced hepatic autophagy are detailed below.
Estrogen-related receptor alpha
Estrogen-related receptor alpha (ESRRA) is an orphan receptor that acts as a transcriptional target of THRs [22]. Research has demonstrated that ESRRA binds to the promoter regions of various autophagy-related genes, including Unc-51-like kinase 1 (ULK1), upon stimulation by TH [22]. ULK1 stands out among ATGs due to its kinase activity, which regulates the recruitment of additional ATGs to the site of autophagosome formation [23].
Forkhead box protein O1
Forkhead box protein O1 (FoxO1) is a key transcriptional regulator of autophagy in the liver [24]. TH has been shown to activate hepatic FoxO1 via deacetylation events mediated by Sirtuin 1 (SirT1), facilitating its nuclear shuttling and transcriptional activity [25,26]. Consequently, FoxO1 likely plays a pivotal role in TH-induced hepatic autophagy.
Transcription factor EB
Transcription factor EB (TFEB) acts as a master regulator of hepatic autophagy, binding to the promoter regions of several autophagy genes and promoting both autophagosome biogenesis and autophagosome–lysosome fusion [27]. Increased TFEB transcriptional activity not only promotes the degradation of bulk autophagy substrates, such as long-lived proteins, but also facilitates the clearance of LDs and damaged mitochondria [27]. This underscores the key role of TFEB in regulating organelle-specific autophagy, including lipophagy and mitophagy. Intriguingly, in animal models of MASLD, TH-induced autophagy and the concurrent reduction in hepatic steatosis have been linked to increased TFEB expression in the liver, suggesting its role in TH-driven selective autophagy [28].
Go to :
THYROID HORMONE INDUCTION OF LIPOPHAGY AND ITS ROLE IN HEPATIC LIPID CATABOLISM
Lipophagy is a selective form of autophagy in which cytosolic LDs, rich in triacylglycerol (TAG), are recognized by autophagosomes. These LDs are subsequently transported to lysosomes, where lysosomal acid lipase degrades the TAGs into free fatty acids (FFAs). The FFAs are then released back into the cytosol [29]. Lipophagy is recognized as a key pathway for TAG degradation in hepatocytes, and its dysregulation is implicated as a major contributor to hepatic steatosis in MASLD in both animals and humans [30]. In research conducted by Sinha et al. [12], TH was found to increase lipophagy in the mouse liver. Notably, when the autophagy-related gene ATG5 was genetically silenced in mice, significant decreases in TH-induced mitochondrial β-oxidation and ketogenesis were observed [12]. These findings highlight the essential role of lipophagy induction in the process of TH-induced lipid catabolism within hepatocytes [12].
Further studies in human hepatic cell lines have shown that TH directly regulates a protein localized to LDs, known as angiopoietin-like 8 (ANGPTL8)/β-trophin/C19orf80 [31]. The induction of this protein appears essential for TH-induced lipophagy [31]. Considering the role of lipophagy in reducing intrahepatic lipid accumulation, its stimulation is thought to decrease the risk of lipotoxicity and prevent lipid-induced hepatic insulin resistance associated with MASLD [32,33]. Thus, the beneficial effects of TH observed in patients with MASLD may be due to TH-stimulated lipophagy (Fig. 1) [34].
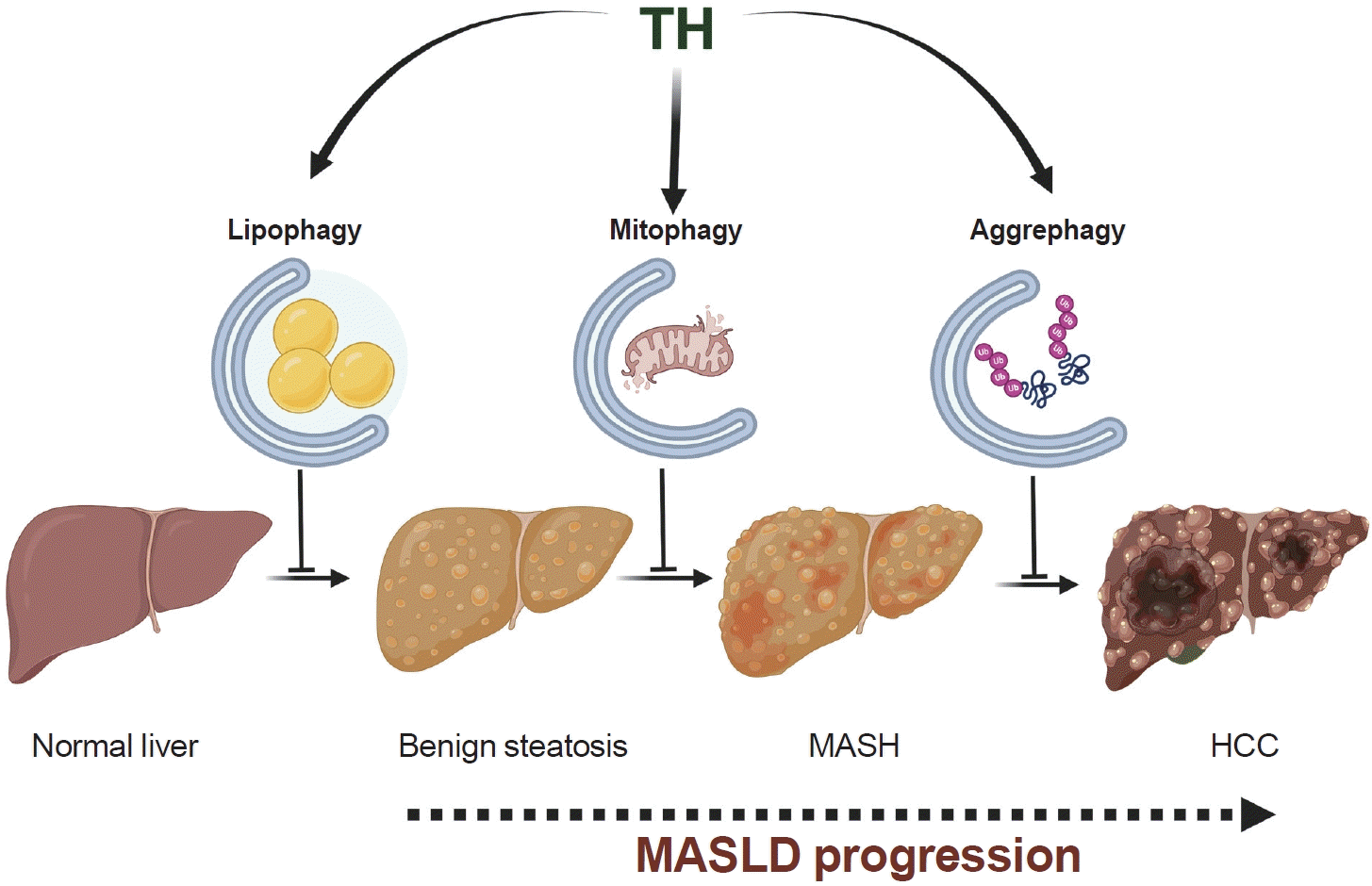 | Fig. 1.Potential role of thyroid hormone (TH)-induced selective autophagy in mitigating metabolic dysfunction-associated steatotic liver disease (MASLD) progression. TH-induced selective autophagy, which includes lipophagy, mitophagy, and aggrephagy, may influence various stages of MASLD progression, from benign steatosis to hepatocellular carcinoma (HCC) development. Specifically, lipophagy may help prevent the accumulation of lipids within hepatocytes, thereby preventing the onset of MASLD. In contrast, TH-induced mitophagy induction may play a pivotal role in preserving mitochondrial health and limiting inflammation, both of which are essential in averting the progression to metabolic dysfunction-associated steatohepatitis (MASH). Finally, TH-induced aggrephagy during MASH could act as a key anti-neoplastic mechanism in hepatocytes. These cells are susceptible to protein aggregate accumulation, which increases proteotoxicity and genotoxicity that can lead to HCC development.
|
Go to :
THYROID HORMONE-INDUCED MITOPHAGY AND REGULATION OF HEPATIC MITOCHONDRIAL HEALTH AND INFLAMMATION
TH induces TAG lipolysis through lipophagy, providing a consistent supply of long- and medium-chain FFAs. These FFAs can be activated by conjugation with coenzyme A (CoA), a reaction catalyzed by acyl-CoA synthetases, and then transported to the mitochondria for β-oxidation. This process increases intracellular adenosine triphosphate production and prevents the accumulation of FFAs, which could lead to lipotoxicity through the formation of reactive lipid species such as ceramides [35]. However, prolonged activation of mitochondrial β-oxidation produces reactive oxygen species (ROS), potentially causing mitochondrial damage and cellular stress [36]. Interestingly, THinduced mitochondrial energetics are associated with a selective autophagic process known as mitophagy [13]. TH-induced mitophagy ensures the elimination of any damaged mitochondria from the pool of energetically active mitochondria before they can release apoptogenic proteins [13]. The induction of mitophagy by TH is linked to ROS-induced calcium–calcium/calmodulin dependent protein kinase kinase 2 (CAMKK2)-mediated AMPK activation [13]. Activation of AMPK leads to the phosphorylation of ULK1, which promotes its translocation to damaged mitochondria. There, ULK1 primes the mitochondria for mitophagic degradation by interacting with mitochondrial proteins such as FUN14 domain containing 1 (FUNDC1) and dynamin-related protein 1 (DRP1) [13,22]. In addition to ULK1, TH has also been suggested to initiate mitophagy through the PTEN-induced kinase 1 (PINK1)-PARKIN pathway [37]. Nonetheless, the induction of mitophagy by TH appears to be crucial for TH-induced mitochondrial energetics, as the loss of mitophagic proteins significantly impairs TH-induced hepatic mitochondrial function, hinders lipid metabolism, and triggers an increase in ROS production [13]. TH also promotes mitochondrial biogenesis in conjunction with mitophagy, which helps maintain a healthy mitochondrial pool within hepatic cells [22].
Additionally, mitophagy has been shown to inhibit activation of the nod-like receptor (NLR) family pyrin domain containing 3 (NLRP3) inflammasome [38]. Considering the role of the inflammasome in the progression of MASLD from benign steatosis to the more severe MASH [38], it is notable that TH-mediated mitophagy has been associated with decreased inflammasome activation in animals fed a MASH-inducing diet [39].
The findings collectively suggest that lipophagy induction by TH may be crucial for combating hepatic steatosis during the early stages of MASLD, whereas mitophagy induction could be more relevant in mitigating oxidative stress and inflammation, which are hallmark features of MASH during the later phases of MASLD progression (Fig. 1). This distinction is particularly important in light of evidence showing impaired mitophagic activity in humans with MASH [40,41].
Go to :
THYROID HORMONE-INDUCED AGGREPHAGY AND DEVELOPMENT OF HCC
MASH, a clinically significant condition within the spectrum of MASLD, poses a substantial risk for the development of HCC. Research has shown a 68% increase in the prevalence of MASH-related HCC between 2010 and 2015 [42]. MASH is characterized by the accumulation of insoluble protein aggregates that contain ubiquitinated proteins and the ubiquitin adaptor p62/sequestosome 1 (SQSTM1) [43]. The presence of p62 inclusions in hepatocytes is a critical marker that differentiates benign steatosis from MASH and is predictive of poor prognostic outcomes for subsequent liver carcinogenesis [43]. Furthermore, p62 accumulation is commonly observed in human HCC [44] and has been linked to defects in a selective autophagic process known as aggrephagy, which is responsible for degrading cytosolic protein aggregates [45]. Interestingly, TH has been demonstrated to prevent diethylnitrosamine (DEN)-induced hepatocarcinogenesis through death associated protein kinase 2 (DAPK2)-driven aggrephagy, which reduces DEN-induced DNA damage and hepatic injury [14]. Specifically, in the livers of mice treated with DEN, TH-induced DAPK2 increases the phosphorylation of p62, which is essential for the clearance of polyubiquitinated protein aggregates [14]. Although the role of TH in reducing MASH-induced HCC has not been specifically studied, it is plausible to speculate that TH-induced aggrephagy may be critical in protecting against the progression from MASH to HCC (Fig. 1).
Go to :
CONCLUSIONS
Over the years, autophagy has been recognized as a crucial cellular process in combating lipotoxicity associated with metabolic diseases, including MASLD [46]. Initially considered nonselective, recent studies have highlighted its precise, selective, and context-specific roles in maintaining cellular homeostasis. At present, calorie restriction and exercise are acknowledged as practical methods to induce protective autophagy in MASLD. However, with the recent approval of TH analogs as the first pharmacological treatment for MASLD/MASH, their role as autophagy modulators in humans warrants further exploration. Preclinical studies have shown that TH is a potent inducer of selective autophagy in the liver, indicating that liver-specific TH metabolites and analogs could selectively trigger autophagy while minimizing off-target effects. For example, the TH metabolite 3,5-diiodothyronine has been found to induce autophagy in the liver, although its precise mechanism remains unclear [28]. Regarding the U.S. FDA-approved TH analog Resmetirom, further research is needed to assess its efficacy as a liver-selective autophagy inducer. Moreover, considering the cell type-specific [47] and stage-specific effects [48] of autophagy during MASLD progression, further mechanistic studies are crucial to optimize the application of TH-induced autophagy in combating MASLD pathogenesis.
Go to :
 XML Download
XML Download