

 PDF
PDF Citation
Citation Print
Print



Highlights
• T cell senescence involves CD57 and KLRG1 expression, with loss of CD27 and CD28.
• Senescent T cells produce inflammatory cytokines, contributing to SASP, notably in diabetes.
• Distinguishing T cell dysfunction as exhaustion or senescence is crucial for understanding mechanisms.
• Further research is needed to investigate the effects of senolytics on T cell senescence.
INTRODUCTION
Cellular senescence is a biological phenomenon characterized by the irreversible arrest of cell division and a state of permanent growth cessation [1]. This process plays a crucial role in various aspects of organismal development, tissue repair, and aging [2]. The key features of cellular senescence include cell cycle arrest, development of a proinflammatory secretome, known as the senescence-associated secretory phenotype (SASP), telomere shortening, DNA damage response activation, mitochondrial dysfunction, resistance to apoptosis, loss of proteostasis, etc. [3].
In addition to an increased incidence and severity of various infectious diseases, the older population also demonstrates a higher prevalence of metabolic disorders [4,5]. The coexistence of heightened vulnerability to infections and an elevated risk of metabolic diseases in the elderly emphasizes the intricate interplay between aging processes, immune function, and metabolic regulation [6,7]. This phenomenon can be attributed to various causes, encompassing anatomical and physiological alterations, an elevated risk associated with hospitalization or invasive procedures, and notably, age-related modifications in the functionality of the immune system. While immunosenescence impacts both the innate and adaptive immune systems, its discernible effects are more pronounced in adaptive immune cells [8].
T-cells are major components and central mediators of the adaptive immune system. T-cell aging refers to the gradual immunometabolic changes that occur in T lymphocytes as individuals grow older [9]. Over the past two or three decades, T-cell senescence has been studied using the advanced tools of genetics, immunology, and molecular biology that have shed light on the fundamental processes in biology [9]. This research has identified numerous senescence-relevant mechanisms and pathways in T-cells. Several key features including thymic involution, accumulation of memory T-cells, shrinkage of T-cell repertoire diversity, impaired proliferative capacity, and telomere shortening, characterize the aging of T-cells, influencing their function and contributing to changes in the immune system [10-12]. Emerging evidence indicated that the altered functionality of senescent T-cells may contribute to the dysregulation of immune responses, leading to infectious diseases and autoimmune reactions. While the concept of T-cell aging has been mainly studied in viral infections and autoimmune diseases, many reviews on this topic have been published [13,14], there are not many review papers yet on the role of T-cell senescence in diabetes and metabolic diseases. Therefore, we summarize age-related changes in T-cells and describe the metabolic and epigenetic landscape of T-cell senescence in the context of metabolic diseases. We also provide an overview of recent works showing how T-cell senescence may modulate the progression of human metabolic diseases. Finally, this review provides insightful perspectives on T-cell senescence as a therapeutic target, as well as a biomarker of metabolic disorders.
CHARACTERISTICS OF SENESCENT T-CELLS
Markers of T-cell senescence
The immune system comprises diverse cell types, each fulfilling specific functions to defend the host against foreign pathogens. T-cells, a subset of white blood cells, play a crucial role in adaptive immune responses. They can be broadly categorized into cluster of differentiation 4 (CD4)+ and CD8+, and further differentiated into specialized cells, necessary for specific biological processes such as facilitating rapid responses to infections, aiding in immunological processes, and eliminating virus-infected cells [15-17]. Following the resolution of immune processes, T-cells undergo apoptosis during the contraction phase to return to homeostasis. However, persistent stimulation by pathogens over an individual’s lifetime can lead to T-cell replication, eventual loss of proliferation capacity, and entry into a state of replicative senescence. Additionally, factors such as cell cycle withdrawal, macromolecular damage, and deregulated metabolism also contribute to T-cell senescence [18].
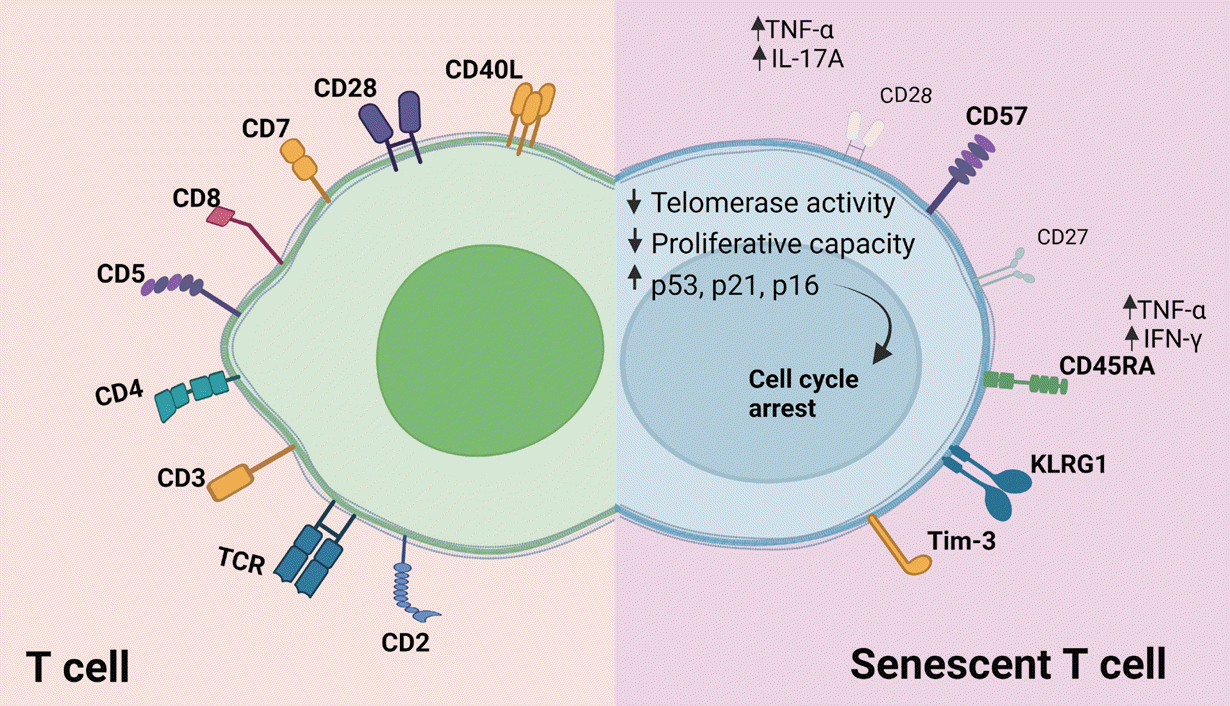
Senescent T-cells exhibit characteristics indicative of cellular senescence. Elevated levels of senescence-associated β-galactosidase (SA-β-Gal) are observed in CD8+ T-cells undergoing replicative senescence, both in vitro and in vivo [19-21]. Analyzing CD8+ T-cells from younger and older donors, the study found that CD8+ T-cells with high SA-β-Gal activity, exhibit features of p16-mediated senescence, and telomere dysfunction-induced senescence. During their differentiation process, senescent T-cells can be distinguished by specific surface markers. Increased expression of CD57, killer cell lectin like receptor G1 (KLRG1) [19], and downregulation of CD27 and CD28 are marked as the phenotypic change related to senescent T-cells [18,20]. Notably, the inactivation of telomerase was found to be associated with the loss of CD28 expression [22]. CD8+ CD28− T-cells contribute to the development of polymyositis, and rheumatoid arthritis, the shorter telomerase, while CD4+ CD28− T-cells stimulate inappropriate immune responses [23,24]. Concurrent with CD28 loss, the expression of CD57 gives rise to a population of CD28−CD57+ T-cells, indicative of terminally differentiated T-cells. This population exhibits higher expression of proinflammatory cytokines compared to CD8+CD28−CD57− and CD8+CD28+CD57− T-cells. Elevated levels of reactive oxygen species (ROS), known to support cellular senescence in fibroblasts, are also found in CD8+ CD28−CD57+ T-cells [25]. However, we need further study to know exactly the role of CD57 expression in T-cell senescence. Investigation of telomere lengths among major subsets based on CD28 and CD27 expression reveals that CD8+CD28−CD27− T-cells exhibit reduced telomerase activity and shorter telomeres compared to CD8+CD28−CD27+ T-cells in humans [26]. Another characteristic feature of senescence is observed in CD27−CD45 isoform RA (CD45RA)+ T-cells, displaying end-stage differentiation characteristics such as the expression of surface CD57, decreased survival, and reduced replicative capacity [27]. Senescent T-cells can also re-express CD45RA, exhibit potent cytotoxic activity, and secrete proinflammatory cytokines such as tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) upon activation [28].
Senescent T-cells are commonly distinguished by specific cell-surface proteins. However, variations in the expression of certain surface markers pose challenges in defining definitive markers for discerning genuinely senescent cells from others. In Fig. 1, we have compiled a summary of T-cell senescence-specific surface markers alongside those of normal T-cells to elucidate the distinctions and unique expression profiles of senescent T-cells.
Exhaustion and senescence: two crucial dysfunctional states of T-cells
Because of their functional similarities, the definitions of senescence and exhaustion can be confusing. However, senescence and exhaustion are two distinct dysfunctional states observed in T-cells, each with its own unique molecular and developmental features [29,30]. The key difference between T-cell senescence and exhaustion lies in the potential for functional recovery. Exhausted T-cells can regain function if the inhibitory signals are blocked or if they are provided with appropriate stimulatory signals. An increasing number of studies have demonstrated the clinical success of immune checkpoint inhibitors, such as anti-cytotoxic T-lymphocyte associated protein 4 (CTLA4) and anti-programmed cell death protein 1 (PD-1)/programmed death ligand 1 (PD-L1) therapies. These treatments can rejuvenate exhausted T-cells, restore their proliferative functions, and enhance the immune response in the tumor environment [31,32]. On the other hand, blocking 5’ adenosine monophosphate-activated protein kinase (AMPK)-TGF-beta activated kinase 1/MAP3K7 binding protein 1 (TAB1)-dependent activation of p38 reversed the proliferative defect in senescent T-cells [33]. Understanding whether T-cell dysfunction in patients is primarily due to exhaustion or senescence not only elucidates the mechanisms driving T-cell dysfunctions in different contexts but also can provide prognostic information and influence treatment strategies.
Senescence is predominantly linked to cell cycle arrest and is often associated with age-related dysregulation of the immune system [30,34]. In contrast, exhaustion arises from prolonged exposure to persistent antigens and is characterized by the upregulation of inhibitory receptors [30]. Exhaustion is frequently observed in patients with infections such as hepatitis C virus and human immunodeficiency virus, as well as in cancer contexts [31,35].
Exhausted T-cells manifest multiple inhibitory receptors and exhibit an altered transcriptional profile, including PD-1, 2B4, Tim-3, T-cell immunoreceptor with immunoglobulin and ITIM domains (TIGIT), glycoprotein CD160, and lymphocyte activation gene 3 protein (LAG3) [29-31,36]. Unlike T-cell exhaustion, senescent T-cells display distinct phenotypic alterations, including elevated expression of SA-β-Gal, increased expression of T-cells senescence surface markers such as CD45RA, CD57, Tim-3, and a downregulated expression of the co-stimulatory molecules CD27 and CD28 [18-21].
Increased expression of LAG3, CD39, and 4-1BB (also called CD137 and tumor necrosis factor receptor superfamily member 9 [TNFRSF9]) has been implicated in dysfunctional antigen-specific CD8+ T-cells within the exhausted tumor microenvironment [37]. Moreover, the loss of functionality and reduced effect of interleukin-2 (IL-2), TNF-α, IFN-γ, and granzyme B (GzmB) are characteristic features of exhausted T-cells [30,35]. Meanwhile, senescent T-cells produce suppressive and proinflammatory cytokines such as TNF, IL-8, IL-6, IL-2, IFN-γ, and TGF-β and IL-10 which constitutes a unique SASP [20].
While the specific molecular processes maintaining the state of T exhaustion cells remain unclear, existing studies suggest that T exhaustion cells acquire DNA methylation programs [38]. Through inhibition of CD28, increasing the expression of p27 and p15, and antagonizing T-cell receptor. PD-1/PD-L1 interactions are known to modulate the metabolic program of effector T-cells, inhibiting the phosphoinositide 3-kinase (PI3K)/Akt/mammalian target of rapamycin (mTOR) and Ras/mitogen-activated protein kinase/ERK kinase (MEK)/extracellular-signal-regulated kinase (ERK) pathways, suppressing IFN-γ secretion in exhausted T-cells [39]. Additionally, PD-1 engagement hampers glycolysis and oxidative phosphorylation (OXPHOS), reducing the number of mitochondria and mitochondrial cristae [39]. Besides, senescent CD8+ T-cells tend to adopt aerobic glycolysis to produce energy instead of using OXPHOS in conjunction with glycolysis. This shift may lead to the accumulation of ROS and mitochondrial dysfunction. Recent research has demonstrated that impaired mitochondria resulting from deficiency in mitochondrial transcription factor A in T-cells, not only accelerate senescence but also trigger various aging-related characteristics, such as neurological inflammation [40]. Within the tumor microenvironment, both natural tumor-derived Treg cells and regulatory T-cells exhibit higher glycolytic capacities and glucose uptake compared to Th17, Th1, and Th2 cells. Treg cells consume more glucose, outcompeting the responding effector T-cells. This heightened glucose consumption triggers ataxia-telangiectasia mutated (ATM)- associated DNA damage and induces senescence in the effector T-cells [41]. Moreover, both senescent and exhausted T-cells exhibit diminished expression of effector molecules, including perforin and GzmB [21]. These findings indicate that T-cell senescence and T-cell exhaustion have different characteristics. Table 1 summarizes the comparison of exhaustion T-cells and senescence T-cells.
Epigenetic landscape of T-cell senescence
In various cellular systems, modifications in DNA methylation, histone alterations, and changes in histone expression contribute to alterations in chromatin structure, which are indicative of the aging process. Such modifications may either directly cause or significantly contribute to cellular dysfunction observed in senescence [42,43].
An investigation into the DNA methylation and gene expression profiles of CD4+CD28− T lymphocytes, utilizing DNA isolated from healthy donors, revealed an association between methylation and cytosine phosphate guanine (CpG) islands. Notably, regions displaying loss of DNA methylation were predominantly located outside CpG islands. The study identified methylated genes associated with T-cell receptor signaling, including protein tyrosine kinase genes, the CD3 complex, and upregulated expression of B cell lymphoma/leukemia type 2 (BCL-2), TYRO protein tyrosine kinase-binding protein (TYROBP), and GzmB. Conversely, demethylated genes were primarily involved in cytokine/chemokine signaling and NK-mediated cell cytotoxicity, incorporating natural killer cell granule protein 7 (NKG7), CD59, granzyme H (GzmH), and GzmB [42]. Senescent CD8+ T-cells undergo functional changes, exhibiting impaired killing abilities due to the loss of GzmB and perforin. Additionally, both senescent CD4+ and CD8+ T-cells express cytotoxicity-associated markers, including granzyme and CD56 [20]. In rheumatoid arthritis, memory CD4+ T-cells demonstrate a senescent CD4+ T cell-like gene expression profile, characterized by the upregulation of GzmB, neural cell adhesion molecule 1 (NCAM1), KLRG1, and the downregulation of PD-1, CD27, and CD28. These findings suggest an elevation of the senescent T-cell phenotype in memory CD4+ T-cells [44].
Examining the relationship between the inflammatory profile, reduced histone expression, and associated replication stress, the reduction of histone expression in young activated CD4+ T-cells resulted in the upregulation of immune and inflammatory responses. This was characterized by increased expression of C-C motif chemokine ligand 3 (CCL3), colony stimulating factor 1 (CSF1), CSF2, interferon regulatory factor 5 (IRF5), and IRF7. Treatment of naive CD4+ T-cell cultures from older adults with the sirtuin 1 (SIRT1) inhibitor attenuated the transcription of these mediators. Significantly, p21 was upregulated, and β-galactosidase activity was observed to be higher in activated CD4+ T-cells from older individuals compared to their younger counterparts. Inhibiting SIRT1 in replicating old human T-cells can restore cell cycle progression and diminish the replication-stress response, suggesting a potential avenue for investigating T-cell senescence [43].
Metabolic reprogramming in T-cell senescence
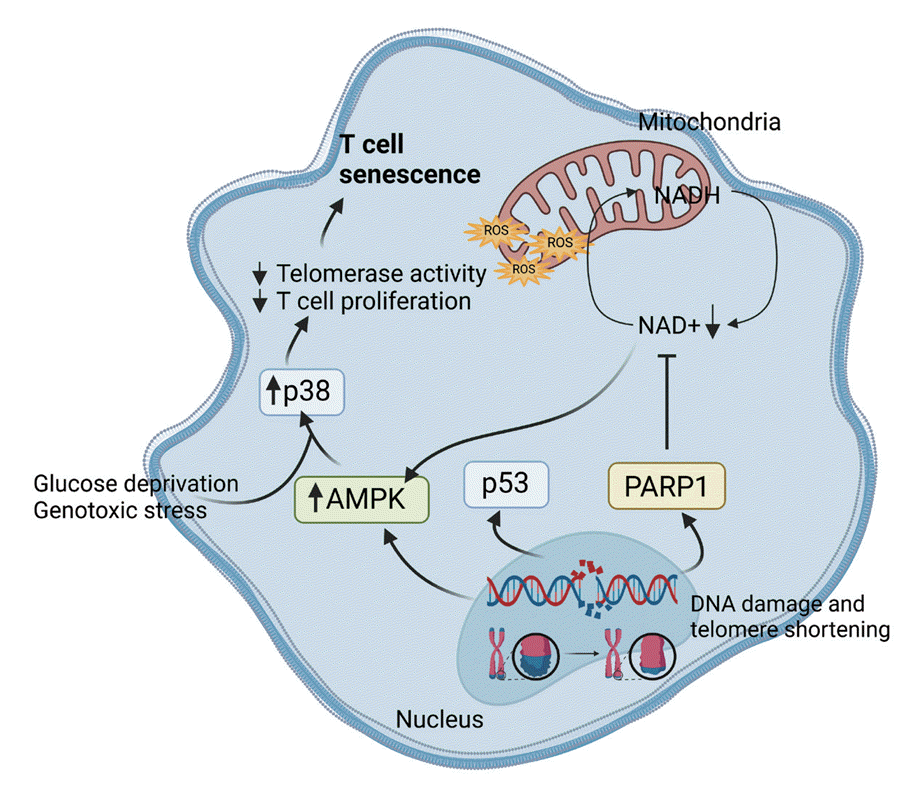
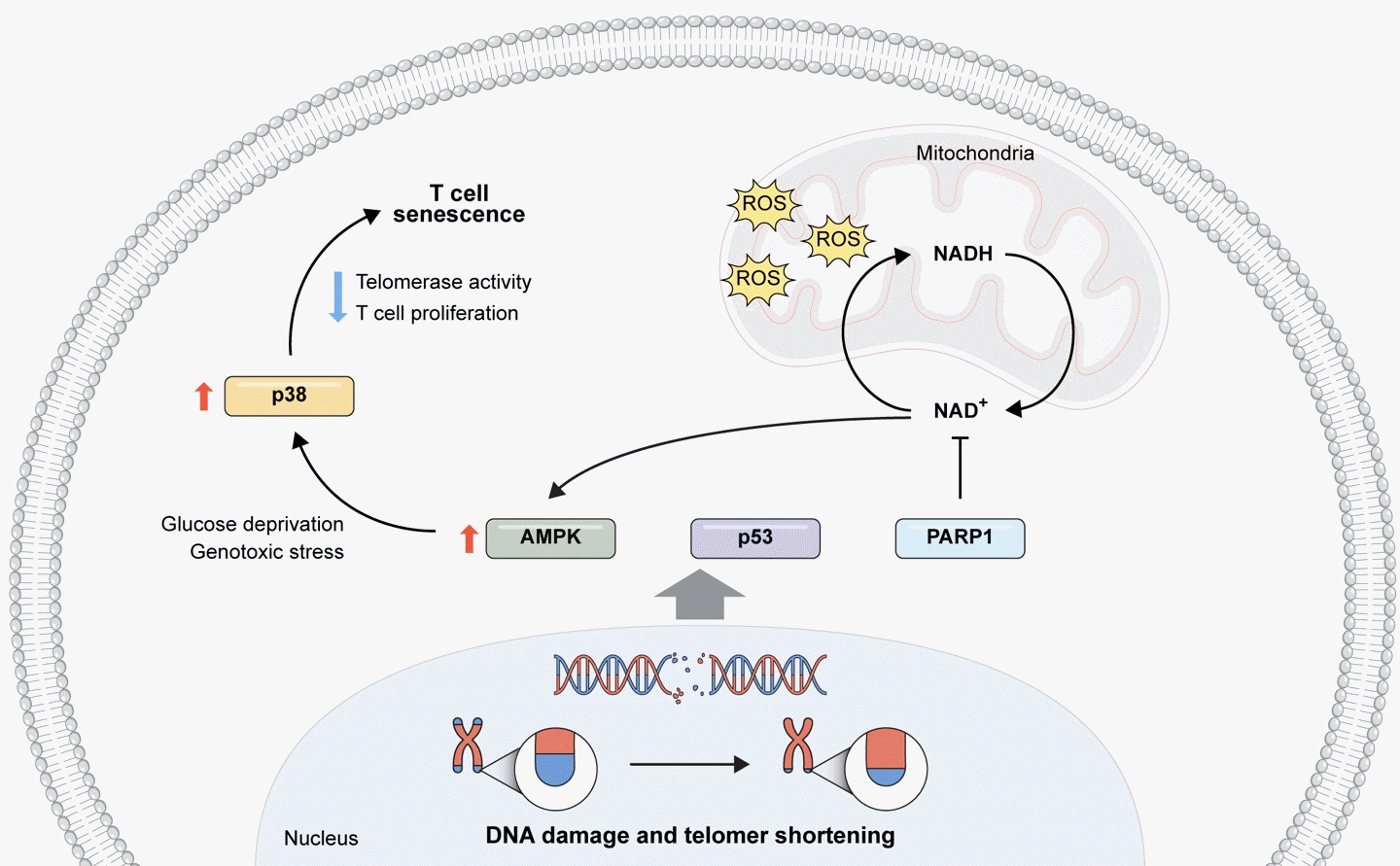
Metabolic regulation plays a pivotal role in cellular senescence, and dysregulated metabolism through various pathways can contribute to the onset of cellular senescence [45,46]. Senescent CD4+CD27−CD28− T lymphocytes exhibit heightened endogenous p38 phosphorylation, activated by the intracellular metabolic sensor AMPK. Specifically, low levels of adenosine triphosphate (ATP) and endogenous DNA damage activate AMPK, inducing constitutive p38 expression in senescent-like CD4+D45RA+CD27− T-cells. Intracellular alterations, such as glucose deprivation and genotoxic stress resulting from p38 activation, lead to a reduction in telomerase activity and the inhibition of T-cell proliferation, representing two key features of senescent CD4+ T-cells [46,47]. The reduction of T-cell receptor (TCR) diversity, critical for the immune system’s ability to recognize a wide range of foreign antigens, characterizes aging T-cells. Analysis of previous data, conducted through nonparametric statistical methods and next-generation sequencing, indicates a two to five-fold decline in the richness of the naive repertoire in healthy elderly individuals compared to TCR β sequences observed in the naive CD4 and CD8 T-cell repertoires of young adults [48]. Additionally, terminally differentiated human CD8+ T-cells (TEMRA) exhibit reduced mitochondrial numbers and a failure to efficiently upregulate OXPHOS following TCR activation. These TEMRA cells also demonstrate elevated levels of p38 MAPK [46].
With its abundance of multiple electron carriers and a robust antioxidant defense system, mitochondria serve as a pivotal hub for regulating the balance between ROS and antioxidants. However, this equilibrium can be disrupted under various circumstances, such as when mitochondrial biogenesis is upregulated without a corresponding increase in antioxidant capacity. This disruption leads to elevated ROS levels, initiating oxidative stress that can inflict damage on mitochondrial components including DNA, proteins, and lipids, thus compromising mitochondrial functionality and efficacy. Consequently, this damage can hinder ATP production and escalate ROS production, perpetuating a detrimental cycle of mitochondrial dysfunction [49]. Conversely, the absence of coenzyme Q10 (CoQ10), either due to pharmacological inhibition or deficiency, exacerbates mitochondrial dysfunction, oxidative stress, apoptosis, and the aging process. Moreover, CoQ10 deficiency has been associated with heightened expression of p21, increased production of SASP, and treatment with CoQ10 notably reduces the number of SA-β gal positive cells [50], shedding light on the significant impact of antioxidants on T-cell senescence. Mitochondrial senescence and dysfunction contribute to inflammation as a feature of aging, particularly observed in aged T-cells or senescent lymphocyte subsets [51]. T-cell response to vaccination requires a delicate balance between the generation of inflammatory effector T-cells and follicular helper T-cells. However, during aging, metabolic interference leading to a shift from glycolysis to mitochondrial respiration disrupts this balance, favoring the differentiation of T-cells into short-lived effector cells. Furthermore, age-related alterations in both the quantity and quality of the T-cell compartment contribute to impaired vaccine responses in older individuals [52].
Maintaining a healthy mitochondrial nicotinamide adenine dinucleotide (NAD) pool and optimal NAD+/nicotinamide adenine dinucleotide+hydrogen (NADH) ratios are essential for normal mitochondrial function. NAD+ serves as a substrate for various redox reactions and as a cofactor in numerous oxoreduction pathways, including the maintenance of mitochondrial membrane integrity and ATP production. Prolonged NAD+ depletion can result in mitochondrial permeability transition, mitochondrial membrane depolarization, and mitochondrial dysfunction, all implicated in poly (ADP-ribose) polymerase 1 (PARP1)-mediated cell death. During aging, the accumulation of damaged DNA activates PARP1, an NAD+-dependent enzyme, leading to NAD pool depletion in a PARP1-dependent manner and subsequent mitochondrial dysfunction [51]. Recent research has identified the involvement of NAD+ utilizing enzyme CD38 in the decline of NAD+ during aging. The expression and activity of CD38 increase during chronological aging, with this upregulation of CD38+ cells partially mediated by the SASP of senescent cells [53]. This suggests a strong link between NAD+ decline during aging and cellular senescence. Consequently, disturbances in the NAD+/NADH ratio and the NAD pool can contribute to mitochondrial dysfunction, initiating senescence and aging. Here, we describe some of the metabolic stress in the context of driving T-cell senescence, specifically summarizing the metabolic reprogramming of these cells in Fig. 2.
DNA and telomere alterations in T-cell senescence
Telomeres, repetitive DNA sequences situated at chromosome ends, play a pivotal role in maintaining genomic stability and safeguarding against the loss of genetic material during cell divisions. As cells undergo division, telomeres gradually shorten, and upon reaching a critically short length, they can trigger cellular senescence or apoptosis, this is one of the characteristics of T-cell senescence [19,35]. Both p21 and p16, two cell cycle inhibitors, contribute to T-cell senescence by inducing cell cycle arrest. Increased expression of p21 leads to inhibition of cyclin-dependent kinase (CDK) activity, p21 can also affect T-cell senescence by regulating DNA damage response pathways. While p16, another CDK inhibitor that specifically inhibits CDK4/6, is implicated in stress-induced senescence. The upregulation is often observed in response to telomere shortening [54,55]. In senescent human T-cells, AMPK triggers the recruitment of p38 to the scaffold TAB1, leading to p38 autophosphorylation. Signaling through this pathway inhibits Tcell proliferation, telomerase activity, and the expression of key components of the TCR signalosome, ultimately driving T-cell senescence [33,46]. Activation of the DNA damage response at telomeres results in the formation of telomere-associated DNA damage response foci (TAFs), recognized markers of cellular senescence in cultured cells and tissues. In humans, TAFs increase with age, independent of telomere shortening, observed in skin melanocytes and CD8+ T-cells, alongside other senescence markers [19]. Defective DNA repair nuclease double strand break repair nuclease (MRE11A) in CD4+ T-cells can lead to telomere damage, and in another study in rheumatoid arthritis patients with low expression of MRE11A showed the increase CD57, p21, and p16 expression [56].
Cytokines from senescent T-cells
Comprehending the specific cytokines implicated in senescent T-cells is imperative for elucidating the intricacies of immune senescence and its implications for health and disease. Senescent T-cells manifest a SASP, marked by augmented production of suppressive cytokines, such as TGF-β and IL-10, alongside proinflammatory cytokines, including IFN-γ, IL-8, TNF, IL-2, and IL-6 [20,30,57].
The heightened senescent phenotype observed in cytomegalovirus (CMV)-specific CD4+ T-cells in healthy donors, relative to other virus-specific T-cell populations, may result from CMV-induced activation of IFN-α by plasmacytoid dendritic cells. This activation inhibits telomerase activity and induces the loss of co-stimulatory molecules in activated CD4+ and CD8+ T-cells. CMV stimulation also leads to elevated concentrations of TNF-α, which not only inhibits both the transcription and translation of human telomerase reverse transcriptase but also interferes with AKT kinase activation while enhancing p38 MAPK activity. These molecular events, previously linked to telomerase activity inhibition, contribute to the distinctive senescent features observed in CMV-specific CD4+ T-cells [27,47].
Senescent T-cells exhibit cytotoxic effects and produce proinflammatory cytokines. Specifically, C-X3-C motif chemokine receptor 1 (CX3CR1)+CD57+CD8+T-cells and CD8+ CD28− T-cells can proliferate in the presence of co-stimulating cytokines, such as IL-15 [22,58]. Studies have indicated that an increased IL-12/IL-10 ratio and enhanced co-stimulatory molecule expression foster a more efficient T-cell response, as dendritic cells can present antigens more effectively. Targeting NK cells to produce IFN-γ and TNF, attracting and activating macrophages [57]. Senescent CD4+ T-cells have been found to secrete substantial amounts of proinflammatory cytokines, particularly IL-6 and TNF-α [20]. Despite their reduced proliferative capacity, senescent T-cells can re-express CD45RA, showcasing potent cytotoxic activity and the secretion of proinflammatory cytokines, such as TNF-α and IFN-γ [28].
T-CELL SENESCENCE IN HUMAN METABOLIC DISEASES
Obesity
Obesity is a chronic and complex inflammatory disorder closely linked to immunosenescence, as suggested by numerous studies over time. In mice, a high-fat diet induced obesity, which increased T-cell senescence in visceral adipose tissue. This was accompanied by an increase in the expression of several markers, including SA-β-Gal, phosphorylation of H2A histone family member X (H2AX) (γ-H2AX), as well as cyclin-dependent kinase inhibitor 1A (Cdkn1a)/Cdkn2b [59]. Shifting our focus to human data, a study of 437 older participants from the Berlin Aging Study II revealed a positive correlation between CD57 expression on central memory CD8+ T-cells and body mass index (BMI) [60]. Additionally, the study demonstrated a positive association between the percentage of effector memory CD45RA-CD27-CD28-CD8+ T-cells, CD57+ effector memory CD8+ T-cells, and homeostatic model assessment for insulin resistance [60]. Moreover, Brunelli et al. [61] classified individuals who had a BMI greater than 30 kg/m2 as obese with type 2 diabetes mellitus (T2DM) in their study. They also identified various senescence markers, including cyclin-dependent kinase inhibitor 2A (p16INK4a) CCR7, CD27, and PD-1 in obese circulating leukocytes [61]. In this study, it was found that CD28 expression decreased while CCR7 expression increased in obese individuals compared to lean subjects [61]. The researchers hypothesized that obesity-related chronic inflammation contributes to T-cell proliferation in successive rounds. Interestingly, a previous study conducted in 2017 found that plasma from obese individuals could cause CD28 expression loss in peripheral blood mononuclear cells (PBMCs) from healthy individuals with normal BMI [62]. This study also highlighted other senescence-related features, such as reduced expression of γ-H2AX and p53 [62]. However, the study found no significant association between obesity and the numbers of senescent CD4+ and CD8+ T-cells, as well as CD45RO+CD57+T-cells within the CD4+ and CD8+ T-cell populations in participants with T2DM [63]. According to the study, there was no significant difference in the numbers of memory CD4+ and CD8+ T-cells producing TNF-α or IFN-γ in participants with T2DM and varying BMI levels. The authors suggest that there is no strong correlation between obesity and T-cell senescence or the inflammatory response in patients with T2DM [63]. There are differences in research results regarding the connection between obesity and T-cell senescence markers. Most of the studies indicate a link between senescent CD8+ T-cells, identified through commonly used surface markers, and obesity. However, no such correlation has been found for CD4+ T-cells in overweight individuals. In individuals with T2DM in the Korean population, there seems to be no correlation between obesity and the presence of senescent CD4+ and CD8+ T-cells, despite using CD45RO+CD57+ as the sole marker of senescence. Therefore, it is necessary to investigate other characteristics of T-cell senescence to support this finding.
Type 2 diabetes mellitus
T2DM is characterized by progressive insulin resistance, pancreatic β-cell secretory deficit, and chronic inflammation. A study conducted with participants from the Diabetes Alliance for Research in England revealed increased levels of CCR7–CD45RA+ senescent subset in both CD4+ and CD8+ T-cells among individuals living with T2DM [64]. Two years later, this group of scientists used CD45RA+CD28–CD27–CCR7–KLRG1 high as the marker for the senescent CD8+ T-cell subset and found that older individuals with T2DM showed an increased expression of the senescent T-cell subset [65]. Additionally, T2DM serum contains inflammatory mediators like C5, IL-8, and serpin E1, which can drive the senescent phenotype in senescent CD8+ T-cells. More specifically, they found that T2DM induces significant changes in the metabolism of senescent CD8+ T-cells, including increased lipid storage, mitochondrial ROS production, reduced fatty acid oxidation, and altered AMPK activity [65]. Furthermore, they found no correlation between the number of medications individuals were taking and the number of senescent CD8+ T-cells. In 2019, a study conducted with participants in England was paralleled by a study involving the Korean population. In the investigation of 805 Korean individuals within the Yonsei Cardiovascular Genome study, researchers examined the association between senescent T-cells and glycemic status [66]. There is a significant increase in the population of CD8+CD57+ and CD8+CD28– T-cells in patients with prediabetes and T2DM when compared to those with normal glucose tolerance [66]. Moreover, there was a notable correlation between fasting plasma glucose levels and the frequency of CD8+CD57+ and CD8+CD28– T-cells in the peripheral blood. Histological evidence analyzed through immunofluorescence showed a higher infiltration of CD8+CD57+ T-cells in the visceral adipose tissues of individuals with prediabetes or T2DM compared to those with normal glucose tolerance [66]. This pattern was similarly noted among individuals with prediabetes, as they displayed heightened levels of CD28−CD57+CD8+ T-cells, whereas there was no notable difference in CD28−CD57+CD4+ T-cells [62]. Additionally, there were significant upticks in the generation of IFN-γ and TNF-α among both senescent CD4+ and CD8+ T-cells [67]. Furthermore, it was discovered that patients with prediabetes had augmented production of perforin in their senescent CD4+ and CD8+ T-cells, indicating a heightened presence of proinflammatory and cytotoxic senescent CD8+ T-cells within this group. In particular, a significantly larger population of GzmB-producing senescent CD8+ T-cells was found in patients with prediabetes, with no significant difference in GzmB expression in senescent CD4+ T-cells between the two groups. When these cells were co-cultured with HepG2 cells subject to insulin treatment, a partial inhibition of insulin-induced suppression of gluconeogenesis by senescent CD8+ T-cells was observed, suggesting a potential impairment of hepatic insulin sensitivity by these cells. Our recent publication reveals a noteworthy relationship between liver fat quantity and CD8+CD45RO+CD57+ T-cells in individuals with T2DM at Chungnam National University Hospital in Korea [57]. Our findings indicate a negative correlation between high-density lipoprotein levels and senescent CD8+ T-cells, while total cholesterol levels were positively associated with senescent CD4+ T-cells in this group. Consistently, senescent T-cells exhibit increased membrane cholesterol accumulation, which contributes to the senescence phenotype of these cells [68]. As summarized in previously published research, cholesterol is directly associated with inflammaging and T-cell senescence. Cholesterol metabolic pathways orchestrate the activation of the NOD-like receptor pyrin domain containing 3 (NLRP3), inflammasome, involving components such as the sterol regulatory element-binding protein (SREBP) cleavage-activating protein (SCAP)-SREBP2 axis, bile acids, and cholesterol itself [69]. Of note, we discovered a connection between T-cell senescence and TNF-α production by senescent CD8+ T-cells and insulin resistance. It is noteworthy that the placenta of the gestational diabetes mellitus group displayed a notable rise in the number of CD28−CD57+CD8+ T-cells [70]. Hence, studies on both English and Korean populations confirm that the frequency of senescent CD8+ T-cells increases with the duration of T2DM.
In a 2011 study conducted in Italy, researchers observed an expansion of CD4+CD28− T-cells in patients with T2DM, which correlated with poor glycemic control [63]. Moreover, glycosylated hemoglobin (HbA1c) was the sole parameter independently associated with the frequency of CD4+CD28− T-cells in T2DM patients without overt cardiovascular disease (CVD) [63]. Research has shown that in individuals with T2DM, the frequency of CD4+CD28− T-cells is positively linked to both HbA1c levels and the duration of the disease [64]. Additionally, in cases of acute coronary syndrome, the frequency of CD4+CD28− T-cells is also notably associated with T2DM [63]. A study conducted 2 years later, which involved a total of 110 diabetes patients (55 with type 1 diabetes mellitus [T1DM] and 50 with T2DM), demonstrated that the frequency of CD4+CD28− T-cells was significantly higher in T1DM patients compared to those with T2DM and the control group. Other studies have also suggested an increase in senescent CD4+ T-cells [64] and senescent CD8+ T-cells [60] in adults with T2DM. It is worth noting that in both studies, cells were classified as senescent solely through the absence of the co-stimulatory marker CD28. Furthermore, a longitudinal analysis in the Veterans Aging Cohort Study demonstrated potentially significant correlations between terminally differentiated effector memory CD4+ (CD45RA+CD57+CD28− or CD45RA+CCR7−) and senescent CD4+CD28− T-cells, and the likelihood of developing diabetes in individuals with human immunodeficiency virus [65]. According to a recent publication, metformin—a commonly used medication for T2DM— has been found to mitigate T-cell senescence [66]. The drug has also been shown to reduce the secretion of IFN-γ while promoting the production of TNF-α in both CD8+ and CD4+ senescent T-cells. As a result, various senescent markers have been observed on both CD8+ T and CD4+ T-cells in T2DM patients. Therefore, several studies suggest that senescent T-cells contribute to the production of proinflammatory cytokines and other components of SASP in individuals with diabetes. These findings suggest that targeting T-cell senescence could be an emerging concept for T2DM immunotherapy. However, specific pathways that elucidate how diabetes induces T-cell senescence in detail have yet to be fully identified. Further studies are required to comprehensively understand the mechanisms underlying T-cell induction in metabolic disorders.
Cardiovascular disease
Numerous studies have already established a connection between the progression of CVDs and the accumulation of senescent T-cells. In this context, we explore the correlation between senescent T-cells and various cardiovascular conditions, including atherosclerosis, myocardial infarction (MI), and hypertension.
As discussed in the section on T2DM, patients with T2DM exhibited a significantly higher presence of CD4+CD28− T-cells compared to those without cardiovascular events [71]. The expanded population of circulating CD4+CD28− T-cells has also been noted in patients with atherosclerotic disease and has been associated with the recurrence of a cardiovascular event in various publications [72-75]. In Austria, a follow-up study involving 107 chronic heart failure patients over 23 months revealed that the expansion of the CD4+CD28− T-cell subset is associated with the severity of chronic heart failure and serves as an independent predictor of mortality in these patients [76]. Additionally, frequencies of CD57+ T-cells in the CD4+ T-cell population were significantly elevated in patients with acute heart failure compared to control subjects [77]. This cell population exhibited a higher frequency of INF-γ- and TNF-α-producing cells compared to the CD4+CD57− T-cell population [77]. In 2004, it was discovered that human heat shock protein 60 is an antigen recognized by CD4+CD28− T-cells in patients with acute coronary syndrome [78]. Four years later, these authors suggested that CD4+CD28− T-cells produce significant amounts of IFN-γ and perforin, contributing to the promotion of atheromatous plaque destabilization in coronary arteries. Mechanistically, the killer Ig-like receptors expressed on CD4+ CD28− T-cells have been implicated in contributing to this proinflammatory phenotype and are responsible for their cytotoxicity [79]. In another study, Delgobo et al. [80] found an exaggeration of senescent CD4+ T-cells capable of infiltrating the heart and promoting myocardial inflammation and stress response, ultimately leading to age-related cardiac dysfunction. Furthermore, the percentage of CD4+CD28− T-cells was also found to be associated with the occurrence of an atherosclerotic vascular event in end-stage renal disease (ESRD) patients receiving a kidney transplant [81]. Senescent CD4+ T-cells were found to be related to almost all cardiovascular events; however, only CD28 and CD57 were utilized as senescent markers in these publications.
In addition, the frequencies of CD57+ and CD28− T-cells within the total CD8+ T-cell population exhibited a significant positive correlation with heart-femoral pulse wave velocity, excluding brachial-ankle pulse wave velocity and carotid-femoral pulse wave velocity [82]. This study indicates that increasing age was significantly associated with rises in systolic blood pressure and the frequencies of senescent CD8+ T-cells [82]. The presence of CD8+CD28− T-cells increases CVD and adversely affects vascular function. Studies have demonstrated accelerated telomere shortening in T-cells of patients with atherosclerosis [83,84] and MI [85,86]. During the progression of atherosclerosis, CD8+CD28−CD27− senescent T-cells within inflammatory vascular walls consistently produce IFN-γ. Moreover, the frequency of CD8+CD57+ senescent T-cells has been correlated with a poor outcome following acute MI [86]. Intriguingly, a higher frequency of senescent CD57+CD8+ T-cells has been observed in patients with acute MI, correlating with cardiovascular mortality 6 months after acute MI [86].
CMV triggers a robust T-cell immune response, contributing to cardiovascular complications. Persistent viral infections, particularly CMV, result in an expansion of the effector/memory T-cells compartment (CD45RO+CCR7− in humans) and an early manifestation of the immune aging phenotype in humans [87]. Human T-cell senescence induced by CMV infection is indicative of cardiovascular mortality in the elderly population [88]. Furthermore, CMV infection is significantly linked to the accumulation of CD28−CD4+ T-cells [89]. In addition, the maladaptive accumulation of CMV-independent CD28− T-cells is positively correlated with cardiovascular death, whereas higher numbers of memory CD8+CD28+ T-cells are associated with overall improved survival [90]. Hence, the significant accumulation of senescent T-cells in CVDs underscores the importance of immunotherapy strategies aimed at their removal, representing an attractive approach to counteract such diseases.
Hypertension
The average number of anti-hypertensive medications, serving as an indirect indicator of the severity of hypertension, was significantly higher in participants with CD28−CD4+ T-cell expansions [89]. Individuals with hypertension display an elevated frequency of CD8+CD28− or CD8+CD57+ T-cells in PBMCs [91]. A higher frequency of CD57+CD28−CD8+ T-cells, along with increased expression of C-X-C motif chemokine ligand 11 (CXCL11), has been reported in hypertensive patients compared to healthy controls, suggesting a role for immunosenescent proinflammatory cytotoxic CD8+ T-cells in hypertension [91]. In a recent study, a high frequency of CD28− CD4+ Tang cells, a specific T-cell subset termed angiogenic T-cells, was observed in hypertensive patients [92]. CD4+ Tang cells exhibit characteristics of senescence with a significant increase in CD57 expression, and a notable decrease in CCR7 and CD27 expression in hypertensive patients. CD28−CD4+ Tang cells may be functionally relevant to the pathogenesis of endothelial dysfunction in hypertension. This effect may occur through the production of high levels of proinflammatory cytokines such as IL-6, IL-17, IFN-γ, and TNF-α [92]. These findings may identify senescent T-cells as potential targets for immunomodulatory therapeutic approaches for hypertensive patients.
Aging
A study published in 1998 revealed that the expanded size of the CD4+CD28− T-cell subpopulation was more pronounced in individuals older than 61 years, constituting approximately 61% of the 200 study participants [93]. In contrast, individuals younger than 40 years infrequently exhibited CD28-deficient CD4+ T-cells [93]. Simultaneously, another American research group conducted a study involving 60 Caucasian volunteers, suggesting elevated frequencies of CD28−CD4+ T-cells in elderly individuals [94]. In the elderly, these CD28-cells can constitute up to 45% of the total CD4+ T-cell compartment [94]. Similar findings, indicating a statistically significant correlation between CD4+CD28− T-cells and age, were also noted [71]. Because loss of CD28 cannot be considered truly senescent, it can be concluded that there is insufficient evidence to establish a clear relationship between senescent CD4+ T-cells and aging.
In 1996, Fagnoni et al. [95] from Italy analyzed T-cells from the peripheral blood of 102 healthy individuals aged 20 to 105 years, revealing an increase in CD28− T-cells in both CD4+ and CD8+ T-cells, particularly prominent in CD8+ T-cells. CD8+CD28− T-cells displayed cytotoxic activity, including reduced antigen-induced proliferation and enhanced production of proinflammatory molecules [95]. This population has been associated with a diminished immune response to pathogens and vaccines in old age [96] and increased mortality in the elderly [97]. The relative telomere length of both CD4+ and CD8+ T-cells was noted to decrease with normal aging [98]. In a study involving 22 participants from the Belgian population, elderly individuals showed significantly higher proportions not only of CD28−CD57+ cells but also of CD28+CD57+ cells. Additionally, CD28+CD57+ cells exhibited the highest expression of p16, p21, Bcl-2, CD95, CD45RO, CCR5, and PD-1, indicating a senescent phenotype [99]. Recent findings also demonstrated the accumulation of senescent CD28−CD57+ CD8+ T-cells with age [25]. Additionally, the accumulation of senescent CD4+ and CD8+ T-cells with age, defined by the markers CD45RA and CD27, was observed [100]. In another study, healthy human donors were categorized into two age groups, with individuals between 57 and 67 referred to as the older group compared to the younger group aged between 23 and 30 years. The proportion of CD8+ T-cells exhibiting high SA-β-Gal activity increased, reaching an average level of 64% in donors in their 60s [19]. These cells displayed features of senescent cells, including telomere dysfunction and p16INK4a-induced senescence. Furthermore, CD57 [101], or KLRG1 [102] expressing CD8+ T-cells also increased with age in humans, although at significantly lower levels compared to the total SA-β-Gal expressing CD8+ T-cell population. Numerous other studies demonstrated an increase in senescent T-cells during aging, exhibiting proinflammatory potential in age-related diseases such as CVDs [103] and T2DM [104].
In a recent study at the First Affiliated Hospital of Sun Yat-Sen University in Guangzhou, China, 88 healthy donors were enrolled and divided into three groups: a young group (<45 years old), a middle-age group (45–65 years old), and an elderly group (>65 years old) [105]. The frequency of senescent T-cells was determined by the expression of CD3+CD8+/CD4+CD45RA+ CCR7−CD27−CD28−CD57+RRG1+. In contrast to previous reports, the study found a significant increase in the frequency of CD8+ senescent T-cells in the middle-aged group compared to both the young and elderly groups. Similar results were observed for CD4+ senescent T-cells, and the proportion of CD8+ senescent T-cells was higher than that of CD4+ T-cells in each age group [105]. Additionally, as noticed above, the study also reported that metformin plays an anti-aging role by reducing the number of senescent T-cells, promoting an increase in telomerase, and enhancing the number of stem T-cells. In another study, Feng et al. [106] explored the relationship between brown adipose tissue (BAT), obesity, and age-related metabolic disorders. The study suggested that senescent S100A8+ T-cells accumulate in BAT with age. The PBMCs collected from both young individuals (aged 16 to 32) and older individuals (aged 56 to 69) showed a significant increase in S100A8+ T-cells in the older group compared to the younger group. Mice that received S100A8+ immune cells exhibited decreased uncoupling protein 1 (UCP1) expression and increased p16 and p21 expression, along with increased beta-galactosidase staining and decreased BAT thermogenesis. Therefore, the study concludes that human S100A8+ immune cells contribute to inducing aging-like BAT dysfunction [106]. Therefore, a new publication points out that aging displays increased senescent T-cells not only circulating in the blood but also present in BAT.
Renal diseases
As discussed earlier, T2DM is closely associated with the accumulation of senescent T-cells. In a study involving 532 adult patients with T2DM, renal function was examined based on albuminuria levels, and immunosenescence features were assessed using flow cytometry. A significant increase in T-cell senescence, indicated by multiple markers such as CD27/CD45RO, CD28, CD127, and CD57, was observed in T2DM patients with estimated glomerular filtration rate below 60 mL/min [104]. The increase of CD8+CD28– and CD8+CD57+ cells was also observed in patients with stage 3 chronic kidney disease (CKD) [104]. Only the CD8+CD28– cells showed a statistically significant increase with albuminuria [104]. Additionally, the study demonstrated that inflammation increases since stage 3 CKD and higher BMI drives the accumulation of CD8+ CD57+ T-cells. A markedly significant proportion of CD28– CD4+ and CD8+ T-cells were found in older ESRD patients [98]. In patients with ESRD, the number of circulating CD4+ CD28– T-cells may considerably increase, comprising more than 50% of the total CD4+ T-cell population [107]. A noteworthy correlation exists between the expansion of circulating CD4+CD28– T-cells and atherosclerotic vascular events in a cross-sectional study involving patients with ESRD [73]. Furthermore, patients with ESRD face a notably high risk of acute atherosclerotic vascular events shortly after kidney transplantation [81]. Furthermore, the percentage of CD4+CD28– T-cells was found to be associated with the occurrence of atherosclerotic vascular events in end-stage renal disease patients receiving a kidney transplant [81]. Conducting a study on children with CKD under the care of Emory Children’s Center and Children’s Healthcare of Atlanta, it was noted that children with CKD undergoing dialysis exhibited a notable increase in CD57+CD8+ T-cells. Additionally, CD28–CD8+ T-cells that were significantly associated with telomere shortening in CKD patients were also elevated compared to the levels in healthy control [108]. Specifically, only in the memory CD4+ T-cell population of ESRD patients, was there a significant increase in the percentage of CD57+ cells. Notably, both young and old patients demonstrated a significantly lower relative telomere length in CD4+ T-cells compared to healthy controls. Interestingly, a statistically significant independent association of CD8+ T-cell relative telomere length with age but not ESRD was noted [98]. In published studies on renal disease, all markers for senescent T-cells used were CD28– or, and CD57. There is no single reliable biomarker for senescent T-cells; therefore, further investigation into additional markers is needed to verify the status of these cells in patients with kidney diseases. Notably, most patients with ESRD receive immunosuppressive therapy after kidney transplantation, which may influence the profiles of T-cell senescence. Therefore, further studies are necessary to understand the impact of immunosuppressive therapy on the percentage of senescent T-cells in these patients.
Liver disease
A significant association was observed between the amount of liver fat and CD8+CD45RO+CD57+ T-cells in participants with T2DM at Chungnam National University Hospital, Korea [63]. In another study, telomere length and telomerase reverse transcriptase activity were analyzed in peripheral lymphocytes within an age-matched cohort comprising 22 patients with non-alcoholic fatty liver disease (NAFLD), 20 with cryptogenic cirrhosis, and 20 healthy individuals. NAFLD is associated with shorter telomere length and an increased number of cells exhibiting hyper-fragmented chromatin, known as senescence-associated heterochromatin foci. Furthermore, the expression of telomerase reverse transcriptase, responsible for adding nucleotides to chromosome’s telomeres to stabilize senescent cells, was significantly higher in individuals with NAFLD [109]. In chronic alcoholics, the CD8+CD57+ T-cell subset may also be associated with chronic infections, which are characteristic of heavy drinkers [110]. Sim et al. [63] also found significantly higher expression of genes related to senescence (e.g., protein tyrosine phosphatase receptor type C [PTPRC], TIGIT, and TNF) and exhaustion (e.g., PDCD1, CTLA4, LAG3, and TNFRSF9) in CD4+ and CD8+ T-cells from participants with nonalcoholic steatohepatitis, suggesting an association between T-cell dysfunction and the progression of liver disease in humans [63]. Therefore, only senescent CD8+ T-cells with CD57-positive markers were found to correlate to liver fat and alcoholic liver disease. Other markers of senescent were also observed, indicating a potential relationship between liver diseases and senescent T-cells. However, further studies are required to investigate a comprehensive range of senescence markers and various stages of liver diseases.
CONCLUSIONS
Upcoming studies will discuss how T-cell senescence research can advance in the context of diabetes and metabolic disorders, and how it can be utilized for predicting and treating diabetes complications. Emerging studies on the subset analysis of human senescent T-cells using single-cell transcriptome analysis will be available shortly. Understanding the mechanisms and consequences of T-cell senescence in diabetes is crucial for developing novel strategies to prevent and treat diabetes complications. Developing biomarkers to predict the risk and severity of diabetes complications, such as CVD, nephropathy, neuropathy, and retinopathy. For example, the frequency and phenotype of senescent T-cells in the blood could be used as indicators of vascular damage, renal dysfunction, or nerve degeneration. Therapeutics development to modulate or eliminate senescent T-cells to improve glucose metabolism and reduce inflammation. Senolytic drugs, such as fisetin and quercetin, could be used to selectively kill senescent T-cells and enhance insulin sensitivity, eventually attenuating the progression of chronic diabetic complications. Alternatively, immunotherapy, such as checkpoint inhibitors or chimeric antigen receptor (CAR)-T cells, have been engineered to target the urokinase-type plasminogen activator receptor (uPAR), which is upregulated in senescence, and could be used to target senescent T-cells and restore immune function. Furthermore, mitochondrial dysfunction related to proteotoxic stress, reactive metabolites such as ROS, ceramides, fatty acids, abnormal glucose levels, and hypoxic conditions may induce T-cell senescence. Consequently, metabolic regulation could be a pivotal strategy for eliminating T-cell senescence in metabolic diseases. BIRB 796, a p38 inhibitor, and other well-known metabolic drugs such as metformin, rapamycin, and glucocorticoids have shown the potential to suppress SASP, reduce ROS production, and increase mitochondrial activity, thereby preventing T-cell senescence [46,111-113]. Effects of environmental factors, such as diet, exercise, stress, and microbiome, on T-cell aging and immunity could be evaluated in humans with T2DM and insulin resistance. A prospective trial demonstrated that hyperbaric oxygen therapy increases telomere length in peripheral blood cells and reduces the number of senescent T-cells [114]. Similarly, physical exercise, which enhances oxygen levels in the blood, helps prevent cellular senescence in circulating leukocytes [115]. Furthermore, comparison between the T-cell aging profiles and responses across different species, genders, and ethnicities may contribute to identifying the common and unique features in humans with metabolic diseases. In Table 2, we provide a brief summary and comparison of T-cell senescence markers across different human metabolic diseases. In conclusion, T-cell senescence is a promising research area that can provide new insights and opportunities for the prevention and treatment of diabetes and its complications. Future studies should focus on elucidating the molecular and cellular mechanisms of T-cell senescence in diabetes, identifying reliable and specific biomarkers of senescent T-cells, and evaluating the efficacy and safety of senescence-modulating therapies in preclinical and clinical settings.
 XML Download
XML Download