

 PDF
PDF Citation
Citation Print
Print



Highlights
• DGAT2 inhibition leads to mitochondrial dysfunction in hepatocytes.
• In the progression of NAFLD to HCC, lipid metabolism, including DGAT2, decreases.
• HCC patients with low DGAT2 are linked to mitochondrial dysfunction and low survival rate.
• The ESRRA-PROX1 axis regulates mitochondrial function in hepatocytes.
INTRODUCTION
Since diacylglycerol O-acyltransferases (DGATs) are the final and committed process for triacylglycerol (TG) synthesis, they are concerned as potential therapeutic targets of steatosis [1,2], with a question that which enzyme is contributing more to diseases progression. DGAT2 and DGAT1 share same name but the only equivalence is acyltransferase activity in TG synthesis [3]. Notably, DGAT2 inhibition exhibited significant decreases of lipid accumulation in liver than DGAT1. Therefore, DGAT2 inhibitor is tested as a drug for non-alcoholic fatty liver disease (NAFLD). Multiple studies in rodents illustrates the potential therapeutic action in DGAT2 inhibition for fatty liver; moreover, the phase two clinical trial of DGAT2 inhibitor was performed in 2021 [4]. Thus, DGAT2 is primarily focused as a TG synthesizing enzyme and the other functions are underestimated.
Chronic cirrhosis develops into hepatocellular carcinoma (HCC). While viral hepatitis is well-known etiology, NAFLD is newly emerging etiology for HCC patients [5]. Hepatitis virus B and hepatitis virus C are known as the major cause of liver cancer for accounting about 50% to 70% of HCC patients. However, the development of vaccine and treatments ameliorates the incidence and prevalence of viral infection are keep lowering [6]. Moreover, liver damages with viral hepatitis reverses with sufficient medication. Therefore, viral hepatitis has chances not to progresses into cirrhosis. Unlike viral hepatitis, NAFLD has no specific medication approved by U.S. Food and Drug Administration, and the prevalence of NAFLD increased up to 25% globally in a decade. Consequently, the portion of NAFLD in HCC patients is robustly increased from 5% to maximum 30% in 2000 to 2020s respectively considering cryptogenic cirrhosis in HCC patients [5,7,8]. Due to high prevalence and lack of treatments, NAFLD is more concerned as a key cause of HCC. However, little is known about the role of lipid metabolism in HCC [9-11].
We focused on the correlation of DGAT2 and mitochondria in HCC. Mitochondrial dysfunction is known to be a hallmark of cancer [12-15]. However, the incidence of how mitochondria is facing impairment are not fully understood. With sharing membrane with endoplasmic reticulum, mitochondria are also considered as a hub for lipid synthesis such as phospholipid and even TG despite the well-known functions; a tricarboxylic acid cycle and β-oxidation [16,17]. Thus, proteins located in mitochondrion is highly related to lipid metabolism. We analyzed the impact of DGAT2 knockdown (KD) on hepatoma cell line and evaluated the cell characteristics compared to those of HCC patients. In this article, we have shown the changes of lipid-related genes in NAFLD patients and HCC patients and the impact of DGAT2 KD on mitochondrion genes via downregulation of estrogen-related receptor alpha (ESRRA) activity.
METHODS
Cell culture
All experiments with cells were processed within 10 passages term. Human hepatoma cell lines, HepG2 and Hep3B, are used in this paper. Media used are Dulbecco’s Modified Eagle Medium (DMEM) with high glucose, glutamine and sodium pyruvate (Thermofisher Scientific, Waltham, MA, USA), supplied with 10% fetal bovine serum (Thermofisher Scientific) and 1% penicillin streptomycin (Thermofisher Scientific). Cell was grown up to 70% to 80% confluency and subcultured into three 100 pi cell culture dish (Sigma-Aldrich, St. Louis, MO, USA). Media was changed for every 3 days.
Experimental design
Basically, overall studies are held with HepG2 cell line. The groups would be HepG2 control shRNA (shCTR) (pLKO.1 shScramble vector) stable cell line and HepG2 DGAT2 shRNA (shDGAT2) (pLKO.1 shDGAT2-TRNC000096) are compared. HepG2 wildtype cell lines are compared with 1 μM DGAT2 inhibitor treated HepG2 cell line. HepG2 shDGAT2 is compared with HepG2 shDGAT2 and ESRRA overexpression model (pCDH-ESRRA overexpression).
Lentiviral stable cell line
Control (SHC002) and shDGAT2 vectors (TRCN0000005195, TRCN0000005196, TRCN0000005197) are purchased from sigma-Aldrich. Lentivirus is produced from HEK293T cell line with transfection of pMDG.2, pRSV, and pMDLg vector with transfer vector. Collected supernatants are filtered with Millex 0.45 μm filter (Sigma-Aldrich). HepG2 cells were subcultured and treated with viral sup and polybrene (Sigma-Aldrich). Transfected cells were selected with puromycin treatments and stored in forzen vials for later use.
Transmission electron microscopy
Transmission electron microscopy (TEM) is used to detect organelles in cell lines. Target cells are fixed for 24 hours with 2% glutaraldehyde-2% paraformaldehyde in 0.1M phosphate buffer (pH 7.4). Specimen is washed and dehydrated and embedded on Poly/Bed 812 kit (Polysciences, Warrington, PA, USA), and electron microscope oven (TD-700, DOSAKA, Japan) at 65°C for 12 hours. The interest region of block is section in 80 nm and placed on copper grid. The section is then stained with 3% uranyl acetate and 3% lead citrate, final specimen is imaged with a TEM (JEM-1011, JEOL, Tokyo, Japan).
Oxygen consumption rate
Oxygen consumption rate (OCR) is measured with Agilent XF seahorse mito stress test kit (Agilent Technologies, Santa Clara, CA, USA). The experiments are followed with manufacturer’s protocol. Briefly, 8,000 cells of HepG2 are plated on XFe96 well plate on the day before the assay. Next day, the cells were rinsed with XF DMEM solution (Agilent Technologies) with carbon source added. Drugs are treated with the final concentration of 2.5, 1, and 2.5 μM for oligomycin, carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and rotenone/antimycin respectively. For DGAT2 inhibitor treated sample, samples were subcultured with 1 μM of inhibitor. For beta-oxidation test, final concentration of 4 μM etomoxir is treated before oligomycin treatments.
Extracellular acidification rates
Extracellular acidification rates (ECARs) are calculated via Agilent XF seahorse glycolysis stress test kit (Agilent Technologies). Protocols are performed as manufacturer described. Briefly, HepG2 cell are plated on XFe96 plate. Drugs are treated with 10 mM, 2.5 μM, and 50 mM of glucose, oligomycin and 2-deoxyglucose respectively.
Cell proliferation assay
HepG2 cell lines are subcultured on 96 well plate with 5,000 cells/well. Media is changed daily, and cell proliferation rate is measured using cell counting kit-8 (CCK-8) kit (Dojindo, Kumamoto, Japan) following the manufacturer’s protocol. Briefly, 10 μL of CCK-8 solution is added on each plate and incubated on 37°C for 1 hour. The plate is then measured with colorimeter to get absorbance rate.
Assay for transposase-accessible chromatin sequencing
Raw fastq files are processed quality check and trimming with Trim Galore. The reads are aligned and paired with Bowtie2, samtools, Picard and shell script.MACS2, and Chipseeker packages are used for peak calling and annotation. The following processes are performed via MACROGEN (Seoul, Korea).
RNA sequencing
Transcriptomic data are collected either from database or lab made libraries. In this article we utilized two RNA sequencing data comparing control HepG2 cell line with HepG2 DGAT2 KD cell line, and control HepG2 cell line vs. HepG2 cell line with DGAT2 inhibitor treated samples. From public database, The Cancer Genome Atlas (TCGA) of liver cancer patients (normal tissue=50, HCC patients=371) data and GSE9632 (NAFLD), GSE135251, GSE1141198, and GSE193084 are analyzed.
HepG2 DGAT2 suppressed models’ RNA is extracted via TRIzol treatment and processed through next generation sequencing. Produced FASTQ files are performed through fastqc and trimmomatic. The reads counts are aligned via HISAT2, Bowtie2, and StingTie. The following processes are performed via MACROGEN. Raw count data are processed to DESeq2 to find differentially expressed genes (DEGs) with false discovery rate (FDR) <0.1, fold change >1.5. Overall gene set enrichments were calculated either Gene Set Enrichment Analysis (GSEA) or gProfiler to find highly significant pathways.
Statistical analysis
All statistical analyses were performed based on the experiment method and characteristics of data. If the data fits in normal distribution, parametric methods are used and if not, non-parametric methods are used. Normality test is performed with Shapiro’s test. For parametric test, Student’s t-test is used. For nonparametric test, mostly Mann-Whitney U test is utilized for calculating significance of correlation between two factors. Also, spearman correlation, long rank test, one-way analysis of variance (ANOVA) and FDR is used for specific methods and are mentioned in the figure legends. For the methods not indicated in following section are described in Supplementary Methods.
RESULTS
Inhibition of DGAT2 mediates mitochondrial dysfunction in HepG2 cell line
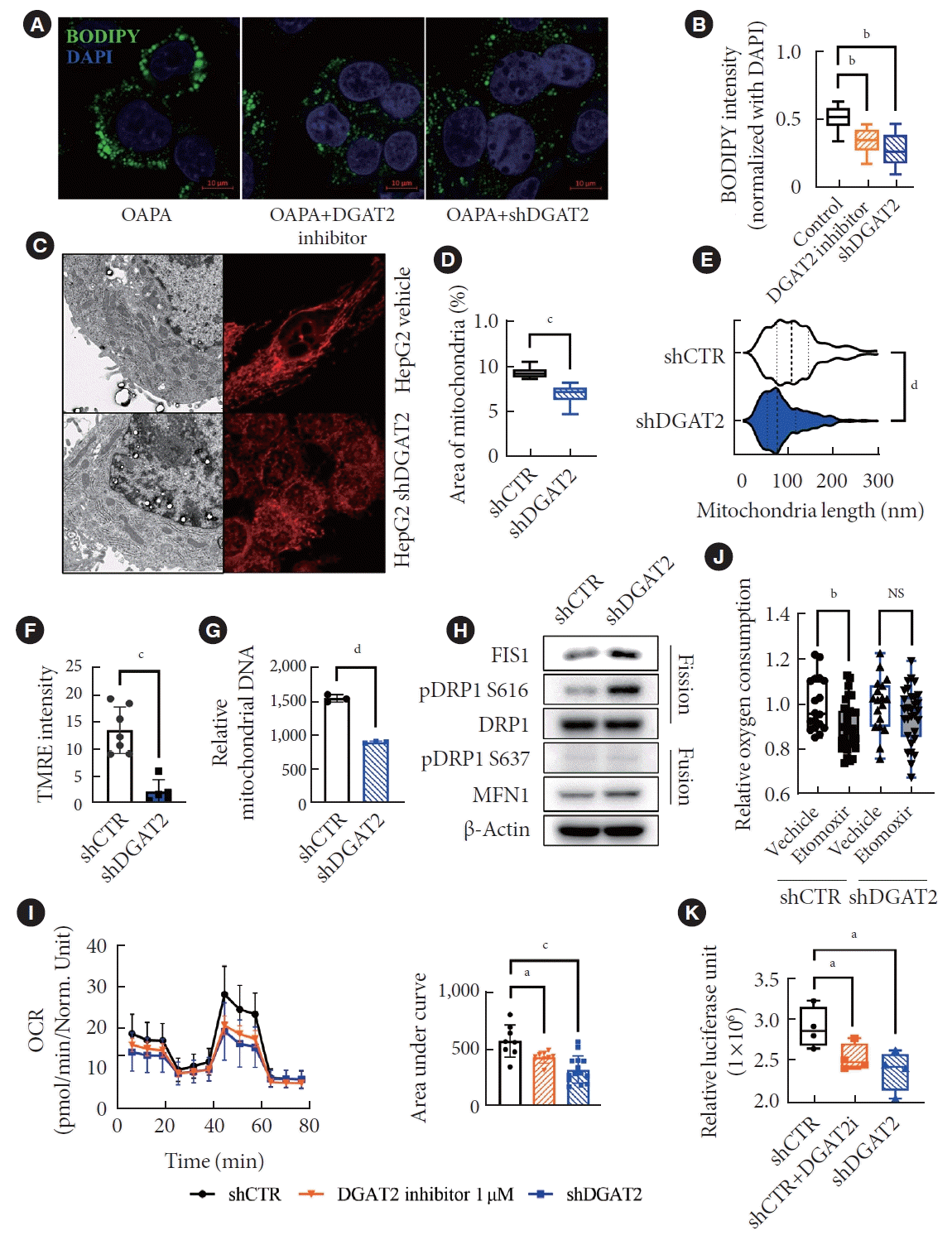
As a family of acyltransferase group, the primary role of DGAT2 is a TG synthesis. We inhibited DGAT2 activity in HepG2 cell line via DGAT2 inhibitor [18], and shDGAT2 (Supplementary Fig. 1A-E). Inhibition of DGAT2 suppresses the production of lipid droplets while 200 mM of oleic acid and palmitic acid are treated (Fig. 1A and B). Along the decrease of lipid droplet, depletion of DGAT2 enzyme markedly diminished mitochondria (Fig. 1C). The relative mitochondria area in cytosol, and the distribution of mitochondrial length suggests significant decrease in its size (Fig. 1D and E). This change is not associated with only morphologies, but the mitochondrial DNA contents and membrane potentials are also decreased (Fig. 1F and G). We assumed the dysregulation in mitochondrial dynamics mediated the morphologies, and were able to detect upregulated fission proteins, fission, mitochondrial 1 (FIS1), and phosphorylation status of Ser616 of dynamin-related protein 1 (pDRP1; Ser616), in DGAT2 KD HepG2 cell line (Fig. 1H) [19]. Consequently, the OCR, especially the maximal respiration rate, is suppressed (Fig. 1I). Moreover, KD of DGAT2 suppressed β-oxidation that etomoxir, carnitine palmitoyl transferase 1 inhibitor, treatments resulted no difference in OCR while control HepG2 resulted significant decrease in OCR (Fig. 1J). Altogether, suppression of DGAT2 activity in HepG2 cell line promotes mitochondrial dysfunction along the decrease of lipid accumulation and eventual decrease of cellular adenosine triphosphate (ATP) level (Fig. 1K).
Lipid accumulation alters upon disease progression in liver
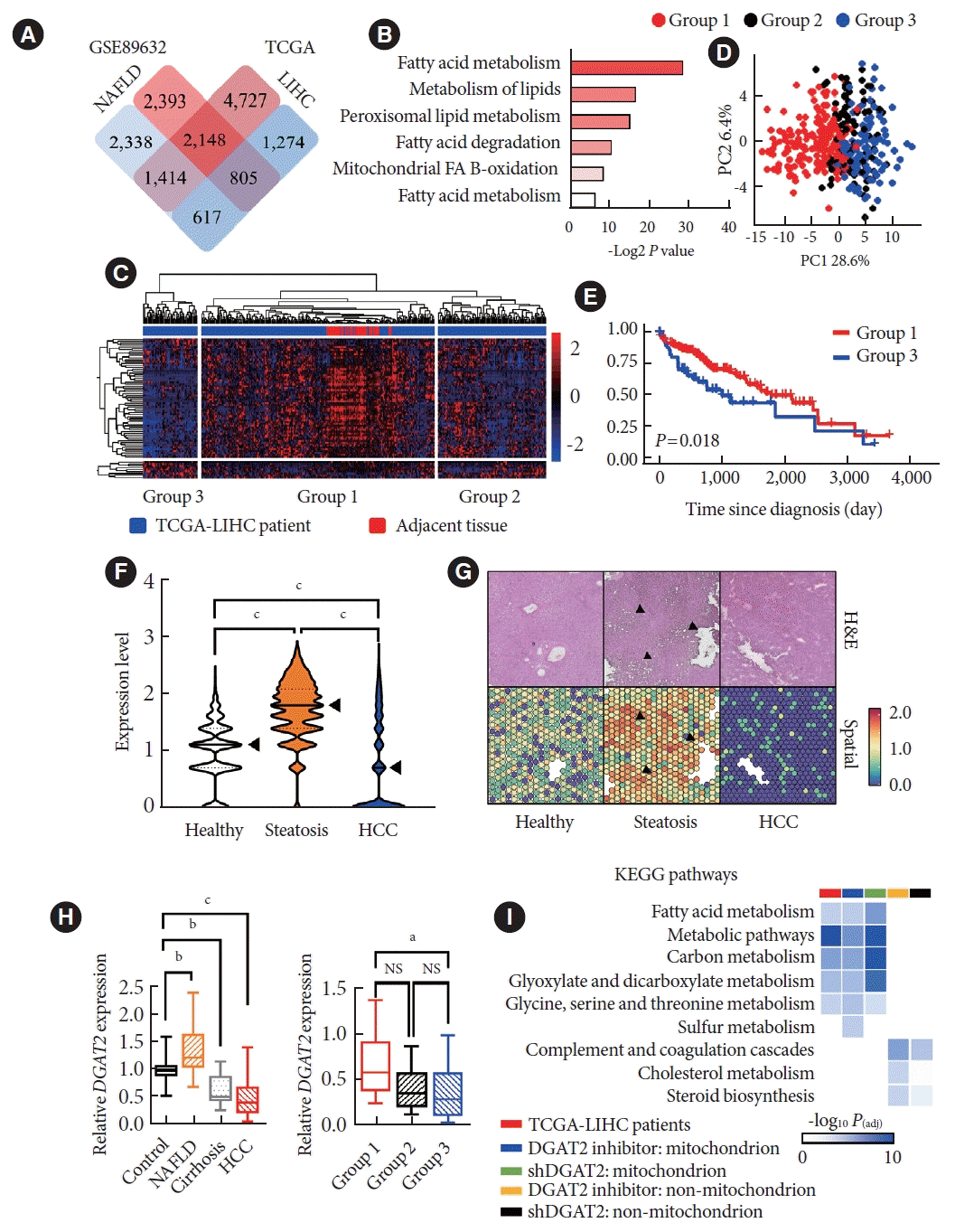
Lipid accumulation in liver upon 5% of total weight are considered as liver steatosis. However, the lipid accumulation is known to decrease in cirrhotic phase. However, the consequences of lipid metabolism in HCC patients are not fully understood. Thus, we analyzed two datasets, NAFLD patients (GSE89632), and HCC patients from TCGA database to find out DEGs in between two cohorts (Fig. 2A). Among four groups of DEGs (Supplementary Fig. 2), 805 DEGs were upregulated in NAFLD patients while downregulated in HCC patients, is the only group to exhibit lipid-related genes (Fig. 2B). This clues that even HCC exhibits suppression in lipid metabolism. Thus, we categorized the patients into different group via spearman correlation based on 73 lipid genes. The heatmap categorized HCC patients into three groups. Group 1 are showing highest correlation with adjacent normal tissue, where group 3 exhibits the lowest correlation with the adjacent tissue (Fig. 2C). The clear separation of group 1 and group 3 in principle component analysis plot suggests metabolic differences in between two, while group 3 is shown to has lower survival rate than group 1 (Fig. 2D and E). Notably, the expression level of DGAT2 is highly correlating with the relationship of NAFLD and HCC patients and gradually decreases with the severity (Fig. 2F and G). The spatial data [20,21] exhibits the DGAT2 expression patterns in liver that the level of DGAT2 is upregulated in the region of ectopic fat in steatosis patients, marked with black arrows (Fig. 2G). Moreover, similar outcome is detected from different cohorts that the expression level of DGAT2 decreases as patients progress into cirrhosis and HCC (Fig. 2H). Next, we have performed bulk RNA sequencing for two DGAT2 inhibition models and were able to find that the correlations of downregulated DEGs in our model and HCC patients come from the gene set; GO:CC mitochondrion (Fig. 2I). Therefore, these data draw the correlation between HCC severity with lipid metabolism and mitochondrial functions, especially with DGAT2.
HCC patients with low expression of DGAT2 face lower survival rate with alteration in energy pathways
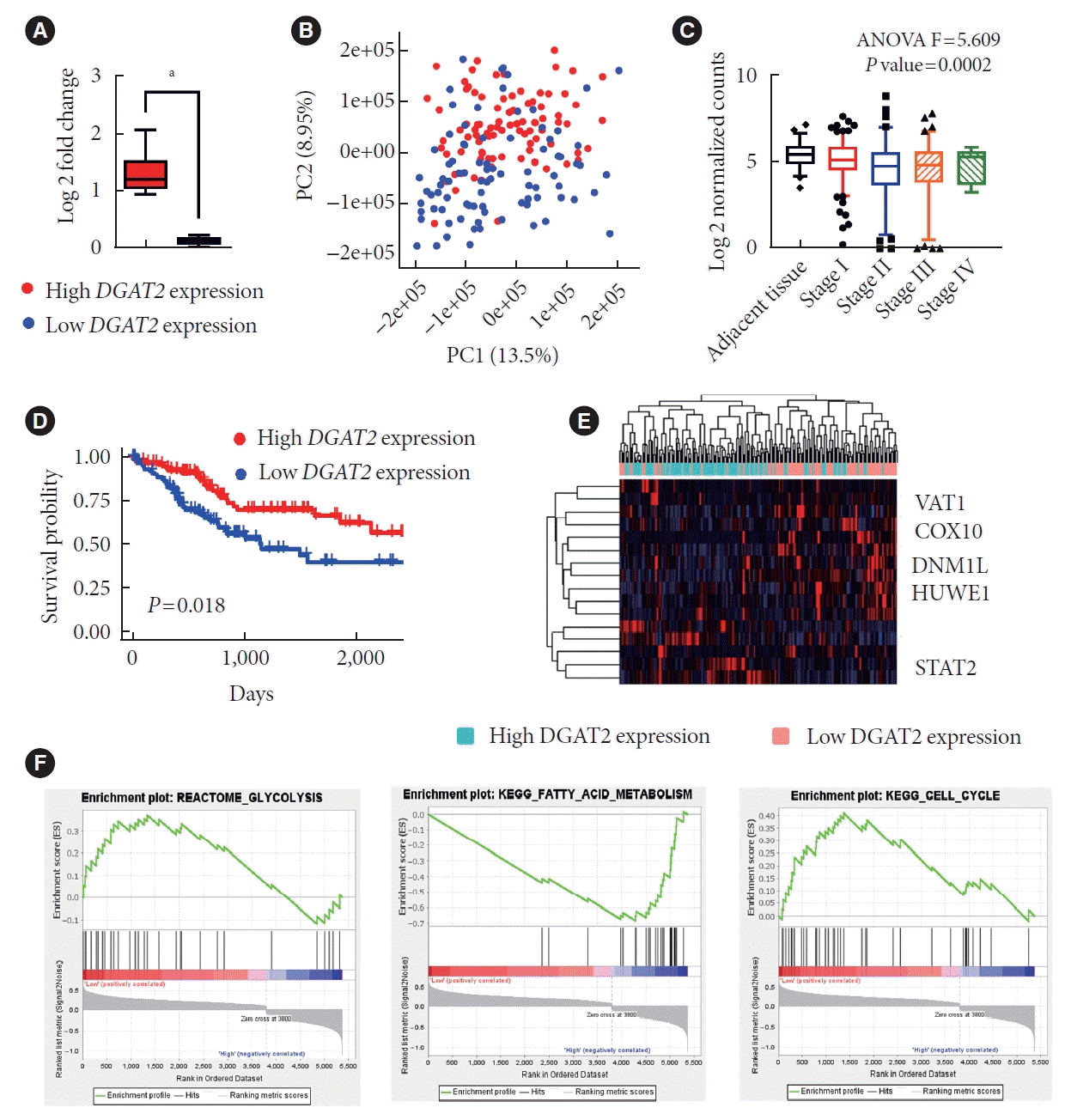
To elucidate the activity of DGAT2, HCC patients are grouped into high DGAT2 expressing group and low DGAT2 expressing group (Fig. 3A and B). Considering that DGAT2 gradually decreases as HCC progresses based on modified tumor-node-metastasis staging (Fig. 3C), patients with low DGAT2 expression tends to show lower survival rate (Fig. 3D). Analysis of DEGs suggest the alteration in the genes regulating mitochondrial dynamics. In low DGAT2 group, fusion inhibiting vesicle amine transporter 1 and dynamin 1 like are upregulated while mitochondrial biogenesis related signal transducer and activator of transcription 2 are downregulated. These imply modification of mitochondrial dynamics (Fig. 3E). Since mitochondrial activity negatively correlates with glycolysis, we run GSEA for probable metabolic changes. The results highlighted upregulation of glycolysis and cell cycle, and downregulation of fatty acid metabolism (Fig. 3F). Therefore, HCC patients exhibited the correlation of DGAT2 and mitochondrial dysfunction.
Mitochondrial dysfunction up regulates glycolysis and promotes cell proliferation
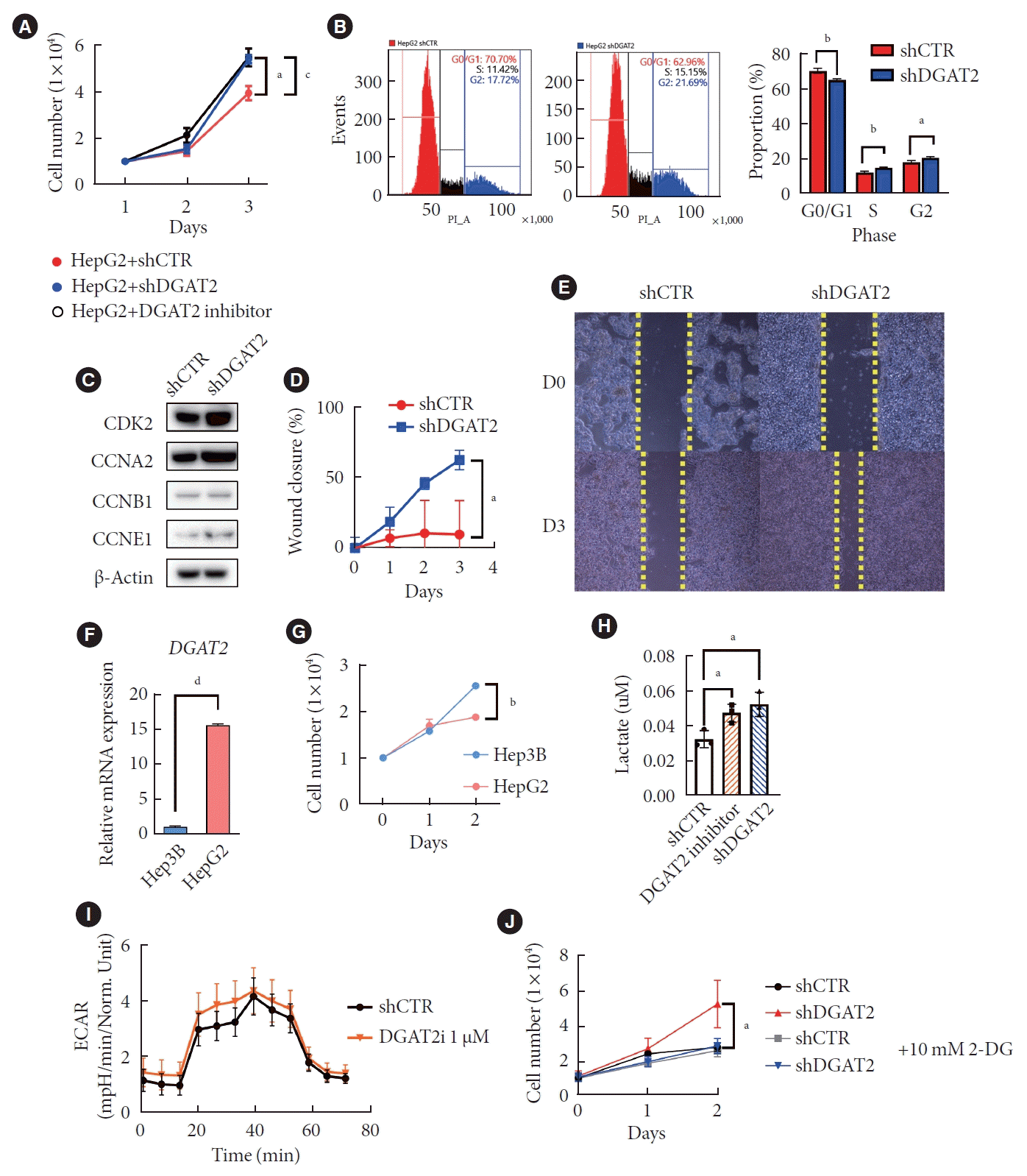
In terms of cancer profile, HepG2 exhibits enhanced cell proliferation rate while DGAT2 activity is suppresses via inhibitor or shRNA (Fig. 4A). Cell cycle analysis with flow cytometry and propidium iodide shows increased S and G2 phase in KD cell line (Fig. 4B). Moreover, protein level of cyclin-dependent kinase 2, cyclin A2, and cyclin E1 are upregulated in DGAT2 KD model (Fig. 4C). Due to upregulated cell proliferation, DGAT2 KD model showed higher speed in wound closure (Fig. 4D and E). Moreover, not only in HepG2 cell line but Hep3B cell line, which expresses low level of DGAT2, exhibits significantly higher cell proliferation rate at day 2 (Fig. 4F and G). This evidence highlights that the DGAT2 mediated mitochondrial dysfunction accelerates cell proliferation, and it can be generalized in other HCC cell lines.
These correlation of DGAT2 expression level and cell proliferation are predicted to be the consequence of upregulated glycolysis in DGAT2 inhibited condition. When DGAT2 activity is suppresses either via inhibitor or shRNA, the final product of glycolysis, lactate, level is increased by 150% of control (Fig. 4H). In addition, ECAR showed the level of glycolysis is increased when DGAT2 is inhibited with 1 μM of PF-06424439 (Fig. 4I and J). As predicted, only DGAT2 KD HepG2 showed suppression in cell growth rate, while it was tolerable for control group. Thus, mitochondrial dysfunction by DGAT2 suppression shifts metabolism toward glycolysis and induces cell proliferation in HepG2 [22].
Estrogen-related response element motif activity is downregulated, which correlates with the DGAT2 expression over liver disease progression
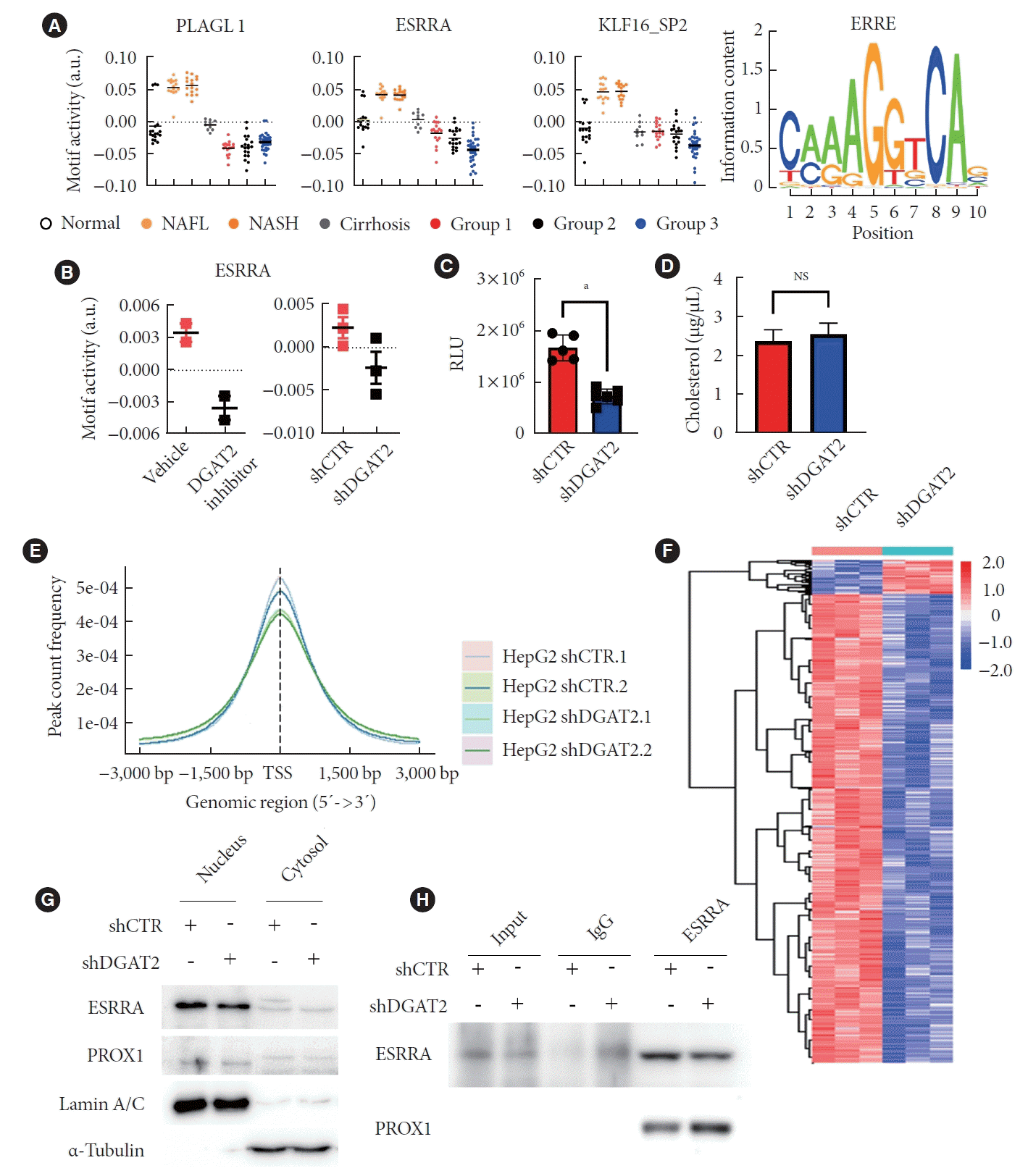
To distinguish a key regulator of metabolic changes, motif activities were calculated with web-based tool, integrated system for motif activity response analysis (ISMARA). The prediction brought three motifs out of top 10 motifs that activated in NAFLD patients but deactivated in HCC patients. Among three motifs, PLAG1 like zinc finger 1, ESRRA, and KLF transcription factor 16, ESRRA fits for the regulating mitochondrial homeostasis and biogenesis (Fig. 5A) [23,24]. ESRRA motif activity from transcriptome of both DGAT2 inhibitor and shDGAT2 model were calculated to be suppresses (Fig. 5B), which confirmed with the luciferase assay we performed. To find motif activity and downstream gene translation. With the insertion of p3x estrogen-related response element (p3xERRE) vector tagged with firefly luciferase, DGAT2 KD model showed half of a relative luciferase activity of control cell (Fig. 5C). However, the only known endogenous ligand of ESRRA, the cholesterol level showed no differences, implying other factors are contributing on the activity of ESRRA (Fig. 5D) [25]. Yet, transcriptome data suggest overall downregulation on the genes with estrogen-related receptor response element (ERRE) sequences and the peak targeted on ERRE are shown to be decreased from analysis of assay for transposase-accessible chromatin sequencing of shDGAT2 model (Fig. 5E and F). Thus, we conducted subcellular fraction to divide nucleus from cytosol. According to Western blot, the amount of ESRRA translocated in nucleus are diminished (Fig. 5G). Notably, immunoprecipitation via ESRRA-antibody from nucleus fraction suggested decreased ESRRA and increased prospero homeobox 1 (PROX1) bound to ESRRA (Fig. 5H). PROX1 is a suppressor of ESRRA; therefore, the transcriptional network of ESRRA-PROX1 deactivates the ESRRA and inhibits downstream gene transcription [26].
Overexpression of ESRRA rescues DGAT2 KD HepG2 from mitochondrial dysfunction
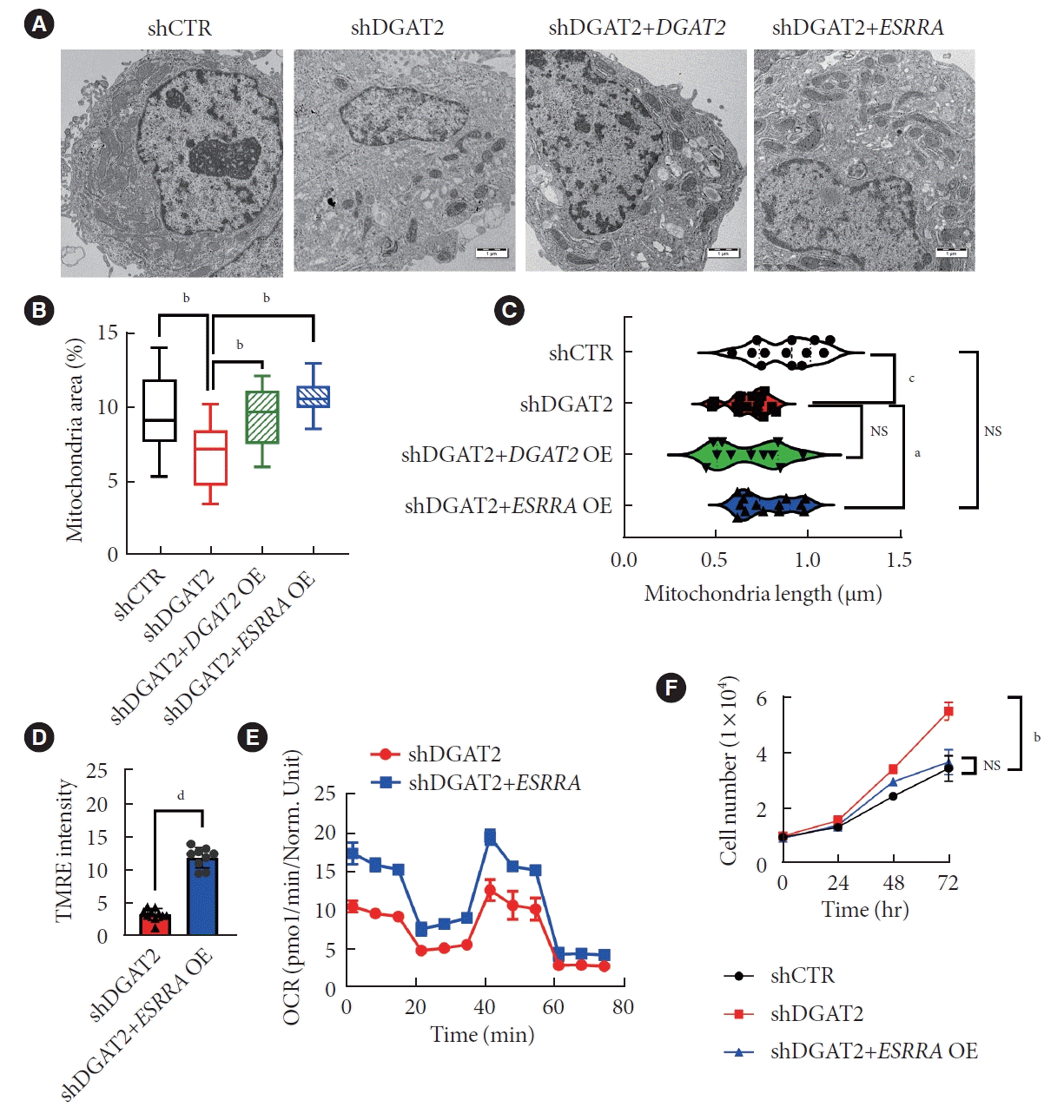
To find out whether downregulated ESRRA is promoting mitochondrial dysfunction, DGAT2 KD HepG2 model is overexpressed with the ESRRA construct. Two days after transfection, cells were performed TEM to assess mitochondrial morphologies (Fig. 6A). Mitochondrial area and length are decreased upon DGAT2 KD and is recued via ESRRA overexpression (Fig. 6B and C). As morphology is rescued, mitochondrial potential is also rescued (Fig. 6D). Following with the increase of OCR via ESRRA overexpression, mitochondrial dysfunction is ameliorated via ESRRA overexpression (Fig. 6E). Moreover, the cell proliferation rate decreased toward control cell compared to the DGAT2 KD model (Fig. 6F). Thus, upregulation of suppressed ESRRA activity reversed mitochondrial function and cell proliferation of HepG2 cell.
DISCUSSION
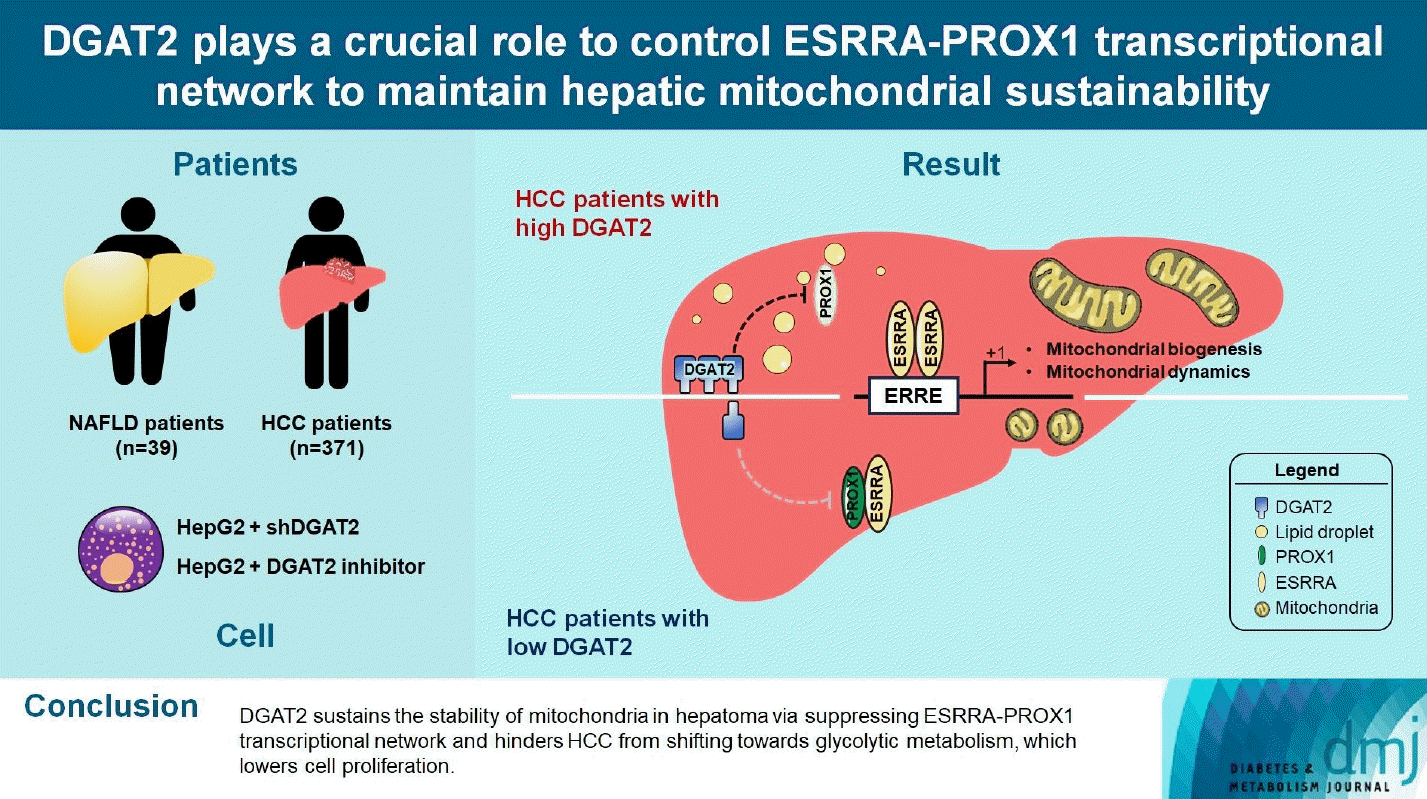
We have presented inhibition of DGAT2, TG synthesizing enzyme, downregulates mitochondrial function via transcriptional network of ESRRA-PROX1. Mitochondria is diminished and cell proliferation is upregulated, which leads to the severity of HCC. Current understanding of mitochondria exceeds its classical role as a cellular powerplant, and further researched as an organelle regulating reactive oxygen species, building blocks, and hypoxia related signals [12]. Although we mainly focused on degradation of mitochondria and classical Warburg effects, upregulated fission could be considered as an advantageous feature for the cancer survival [27]. Fusion is a process of mitochondria to mix damaged mitochondria with healthy one for diluting the damages. Upregulated fission over fusion causes increased stress and promotes mitophagy, which produces energy and biomolecules for cell growths [28]. In TEM of DGAT2 KD cell, upregulated mitophagy is detected, which implies the possible sources for cell proliferation and autophagosome markers are elevated (Supplementary Fig. 3).
While DGAT2 links NAFLD and HCC, it is hard to specify the consequences of a NAFLD on HCC since each patient possesses multiple etiologies. Based on TCGA database of Liver Hepatocellular Carcinoma (LIHC) patients, the number of patients with single etiology are seven for NAFLD. Moreover, usually patients are categorized as non B viral and non C viral (NBNC), which is not actually a NAFLD patients but also includes type 2 diabetic patients and alcoholic fatty liver diseases. It could be the reason but the DGAT2 expression level is not distinguishable according to the etiologies (Supplementary Fig. 4A). The correlation of DGAT2 and other clinical factors from TCGA-LIHC sample suggests higher DGAT2 expression in female; however, no correlation is detected with body mass index (Supplementary Fig. 4B and C). Several studies suggest the correlation of liver fibrosis above F3 are highly correlated with the prevalence of HCC [29]. Notable point is that expression level of DGAT2 not only correlates with NAFLD and HCC, but also negatively correlates with fibrosis level (Supplementary Fig. 4D and E). Therefore, further studies are required to determine when, and which factors regulate the expression of DGAT2 in liver disease [30].
One clue from TCGA data exhibits possible factor for DGAT2 downregulation. Analyzing 50 paired HCCs with corresponding adjacent tissue points that average DGAT2 expression level of adjacent tissues does not represent those of paired cancer; rather, the expression level seems to change during tumor formation (Supplementary Fig. 5A). To figure out possible factors regulating DGAT2 expression, methylation, copy number variant and mutation were evaluated. While copy number variants have no differences, methylation on CpG island of DGAT2 were significantly higher in low DGAT2 expressing group (Supplementary Fig. 5B and C). For mutation, although DGAT2 mutation is known to promote Charcot-Marie-Tooth disease [31], DGAT2 is silently mutated in only two patients out of 371 patients (Supplementary Fig. 5D). It can be implied that HCC obtain methylation during tumor formation, which determines the expression level of DGAT2.
In this paper, we proposed the downregulation of ESRRA as a driving force for mitochondrial dysfunction; however, the direct connection of DGAT2 and ESRRA is still in vague. Probable change from DGAT2 inhibition is the metabolite changes, especially diacylglycerol (DAG). The assay of DGAT2 KD cell line suggest no significant increase in DAG concentration, rather exhibits slight decreased concentration, correlates with increased concentration of phosphatidic acid (PA) level (Supplementary Fig. 1F and G) [32]. DAG is converted into PA via diacylglycerol kinase and used as a derivative of plasma membrane. On the other hand, protein kinase C (PRKC) family has C1 domains for DAG binding for its activation [33-35]. Among PRKC family, PRKC epsilon is downregulated, and it is known to activate ESRRA via phosphorylation on T106, S110, and T124 [36]. Likewise, the cofactor of ESRRA needs further evaluation. PPARGC1A (PGC1A) is one well-known coactivator of ESRRA, while PROX1 is a corepressor of ESRRA. The protein level of PGC1A is significantly downregulated upon DGAT2 KD. In contrast the expression of PROX1 is upregulated (Supplementary Fig. 6A). Moreover, the KD of PROX1 suppressed cell proliferation of DGAT2 KD cell (Supplementary Fig. 6B).
Immune response in tumor microenvironments regulates the growth and survival rate of tumor and immune evasion is considered as a key factor in cancer hallmarks. Little is known about the correlations between DGAT2 and immune function in cancer. Moreover, the controversy perspectives of immune response in NAFLD and HCC make it harder to comprehend that in NAFLD, anti-immune functions are considered as a positive marker where in HCC, pro-immune functions are considered as safer. Based on immune signature, pro- and anti-immune activities are scored [37]. The enrichment score suggests both pro-inflammatory signature, naïve and cytotoxic signatures, and antiinflammatory signature, exhaustion and regulatory T-cell signatures, are upregulated in low DGAT2 group (Supplementary Fig. 7A). Therefore, considering single factor make it hard to interpret the correlation of DGAT2 and immune function. However, comparing the cytotoxic signature with exhaustion signature, only low DGAT2 group exhibits significantly increased exhaustion signature than cytotoxic signature (Supplementary Fig. 7B).
Metabolism shift in the cancer is an essential adaptation for cancer survival. However, the role of lipid in HCC is yet to be clarified due to diversity of lipid categories. We revised HCC in terms of lipogenesis and how mitochondrial function be modulated via DGAT2 and suppresses cancer cell proliferation.
 XML Download
XML Download