

 PDF
PDF Citation
Citation Print
Print



Introduction
Gastric cancer is the third leading cause of cancer-associated mortality worldwide [1]. Most gastric cancers are sporadic. Atrophic gastritis, Helicobacter pylori infection, smoking, consumption of salted food, and a history of gastric surgery are known risk factors for gastric cancer [2-6]. Familial aggregation has been found in approximately 5%-10% of patients with gastric cancer. Twin studies have revealed a higher concordance rate for gastric cancer in monozygotic twins than that in dizygotic twins, suggesting a genetic component in the development of gastric cancer [7,8]. Ethnic differences in the incidence of gastric cancer also suggest a genetic influence [9]. However, not all familial cancers have identified genetic etiologies. Only 1%-3% of all gastric cancer cases correspond to hereditary gastric cancer for which a genetic cause has been identified [10].
Hereditary diffuse gastric cancer (HDGC) is one of the representative syndromes whose genetic causes have been identified. The first reported causative gene for HDGC was CDH1, which was discovered in 1998 by Guilford et al. [11] in a study conducted in New Zealand and Australia. CDH1 is located on chromosome 16q22, and encodes the transmembrane protein E-cadherin. E-cadherin mediates intercellular adhesion and establishes and maintains the integrity and structure of epithelial tissue. The adherens junctions formed in this manner are critical for proper tissue organization [12]. Decreased E-cadherin expression impairs cell adhesion and increases cell motility. This allows cancer cells to detach from the primary tumor and invade surrounding tissues, leading to metastasis [13].
CTNNA1 is another causative gene of HDGC. In 2013, Majewski et al. [14] conducted a study on a HDGC family without CDH1 mutations and revealed CTNNA1 to be another causative gene in HDGC. CTNNA1 is located on chromosome 5q31 and is responsible for coding the cytoplasmic protein, α-E-catenin. Similar to E-cadherin, the main function of α-E-catenin was to link the cadherin-based adhesion complexes at the cell membrane to the actin filaments in the cytoskeleton. It acts as a bridge between the intercellular domain of E-cadherin and the actin cytoskeleton, facilitating the transmission of mechanical forces and stabilizing cell-cell adhesion [15]. Impairment of α-E-catenin function caused by CTNNA1 mutations can disrupt intercellular adhesion and compromise tissue organization. This disruption contributes to cancer progression and metastasis [16].
CDH1 or CTNNA1 mutations have been identified in only 40% of HDGC families, and for the remaining families the factors driving susceptibility remain unknown. This study aimed to identify new pathogenic variants of HDGC and validate their effects on protein expression in new pathogenic mutants.
Materials and Methods
1. Study participants
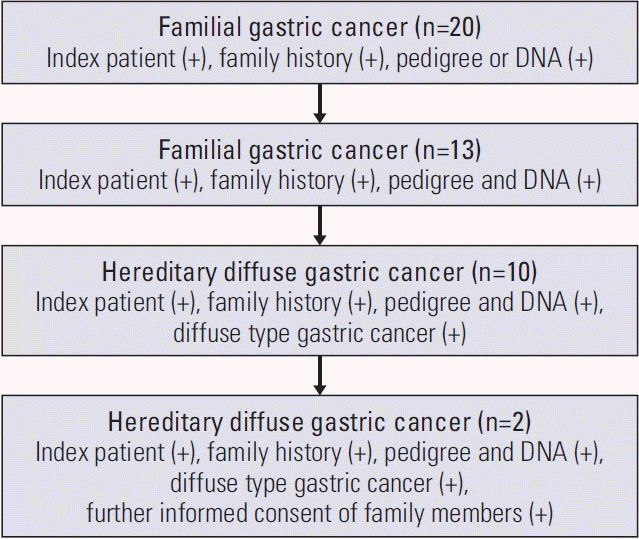
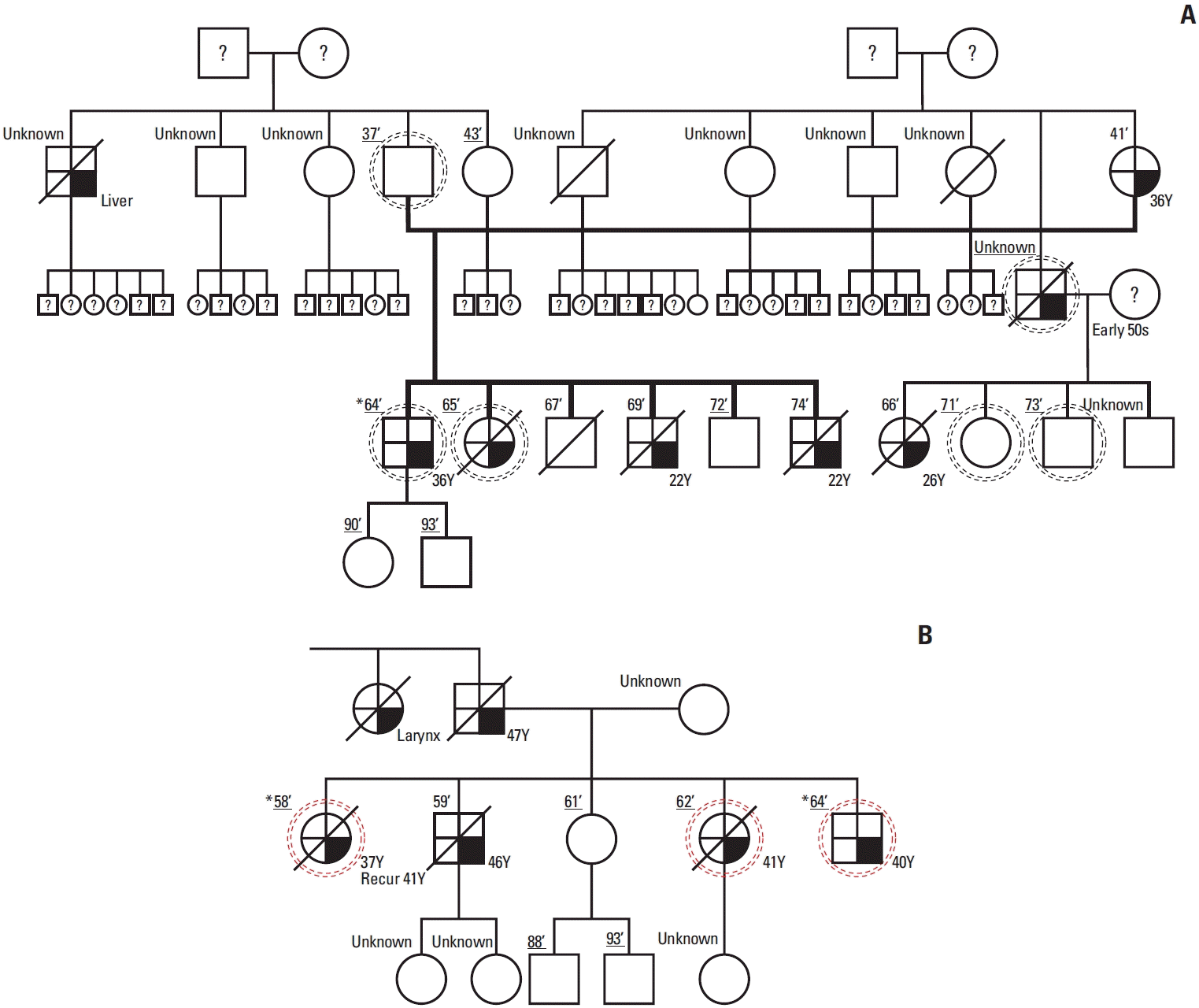
Since 2006, we collected pedigree data of patients who underwent surgery for gastric cancer at Seoul National University Hospital and whose family history met the diagnostic criteria for familial gastric cancer; two first- or second-degree relatives with gastric cancer before the age of 50 years or three first- or second-degree relatives with gastric cancer independent of age. DNA was also extracted from whole blood samples of patients and their relatives. Among these 20 families, both the pedigree and the DNA extracted at that time were available for 13 families, and 10 families met the diagnostic criteria for HDGC. Informed consent for further genetic analysis was obtained from two families included in this study (Fig. 1).
2. Whole genome sequencing
To identify candidates for new pathogenic germline mutations in HDGC, nine blood samples from the index patients and their family members with or without gastric cancer were sequenced using whole genome sequencing (WGS) (Fig. 2). Sequencing reads were aligned to the reference genome GRCh37 using Burrows-Wheeler Aligner (BWA). Variants were called using HaplotypeCaller from the Genome Analysis Toolkit (GATK) and all processes followed the GATK best-practice guidelines. Using WGS data, we detected three candidate genes with germline mutations, EPHA5, MCOA2, and RHOA. Among these, RHOA was selected based on its high frequency of mutations reported in gastric cancer.
3. In-silico analysis
We conducted extensive in-silico analysis to explore the potential impact of the newly identified final candidate on protein function and pathogenicity. Tools such as Sorting Intolerant From Tolerant (SIFT), Polymorphism Phenotyping v2 (PolyPhen-2), Mutation Taster, Combined Annotation Dependent Depletion (CADD) score, Protein Variation Effect Analyzer (PROVEAN), and I-Mutant Suite were used.
4. Whole exome sequencing
To identify RHOA mutations in cancer tissue, we extracted DNA from the formalin-fixed paraffin-embedded tissue block of the two patients whose whole blood samples were previously used for WGS and whose tissue was stored in our institution. Whole exome sequencing (WES) of the extracted DNA was performed.
5. Validation of candidate variant and functional enrichment analysis
To determine whether RHOA mutations could lead to cancer, we designed a functional validation study. We chose cell lines without CDH1 mutations and with low RhoA expression levels. MKN1, SNU216, and SNU668 cells were grown in Roswell Park Memorial Institute 1640 (RPMI 1640, Hyclone, Logan, UT) medium supplemented with 15% fetal bovine serum (Invitrogen, Carlsbad, CA) and 1% penicillin-streptomycin (Invitrogen). All cells were maintained in a humidified incubator at 37°C and 5% CO2. Using these cell lines, we performed site-directed mutagenesis targeting RHOA and overexpressed RhoA. After confirming the RhoA expression pattern in the mutant, we analyzed the change in Rho-ROCK signaling pathways in which the RhoA protein is involved using a RhoA pull-down assay to explore the potential role of RHOA mutation in regulating cellular function.
6. Candidate variant in other families
After functional validation of the germline mutation in the RHOA, we performed WES on the DNA of the index patients from 12 other families who met the diagnostic criteria for familial gastric cancer to determine whether germline mutations in the RHOA were present. None of these patients had mutations in CDH1 or CTNNA1, and the causative gene was not identified.
Results
The mean age at gastric cancer diagnosis among the index patients from 13 families, for which both DNA extracted from whole blood samples and pedigree were available, was 36.8 years, and 76.9% of these patients presented with diffuse-type gastric cancer (Table 1).
There were no CDH1 and CTNNA1 mutations in the two families who provided blood samples of the patient and family members for DNA extraction and WGS (Fig. 2), and the same germline mutation was confirmed among family members in only one family (Fig. 2B, right). Among the three candidate germline mutations of HDGC, EPHA5, MCOA2, and RHOA, RHOA with the most reports related to gastric cancer was selected as the final candidate. This mutation was identified at position 49399952 on chromosome 3, according to the hg19 genomic build, where the reference allele is G, and variant allele is A (chr3:49399952G>A). The variant was R129W, which is present in the insert domain of the RhoA protein [17].
The results from the in-silico analysis to predict the pathogenic effects of the variant suggest that the RHOA mutation (R129W) would induces changes in protein function (Table 2).
To identify the RHOA mutation (R129W) in the cancer tissue, we extracted DNA from the formalin-fixed paraffin-embedded tissue blocks of two patients whose (1) germline mutation of RHOA (R129W) was confirmed in the result of WGSs using DNA extracted from blood samples and (2) cancer tissue was stored at our institution. The same RHOA mutation (R129W) was identified in both DNA samples from tissue blocks of the two patients.
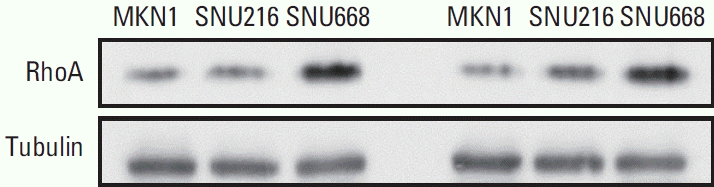
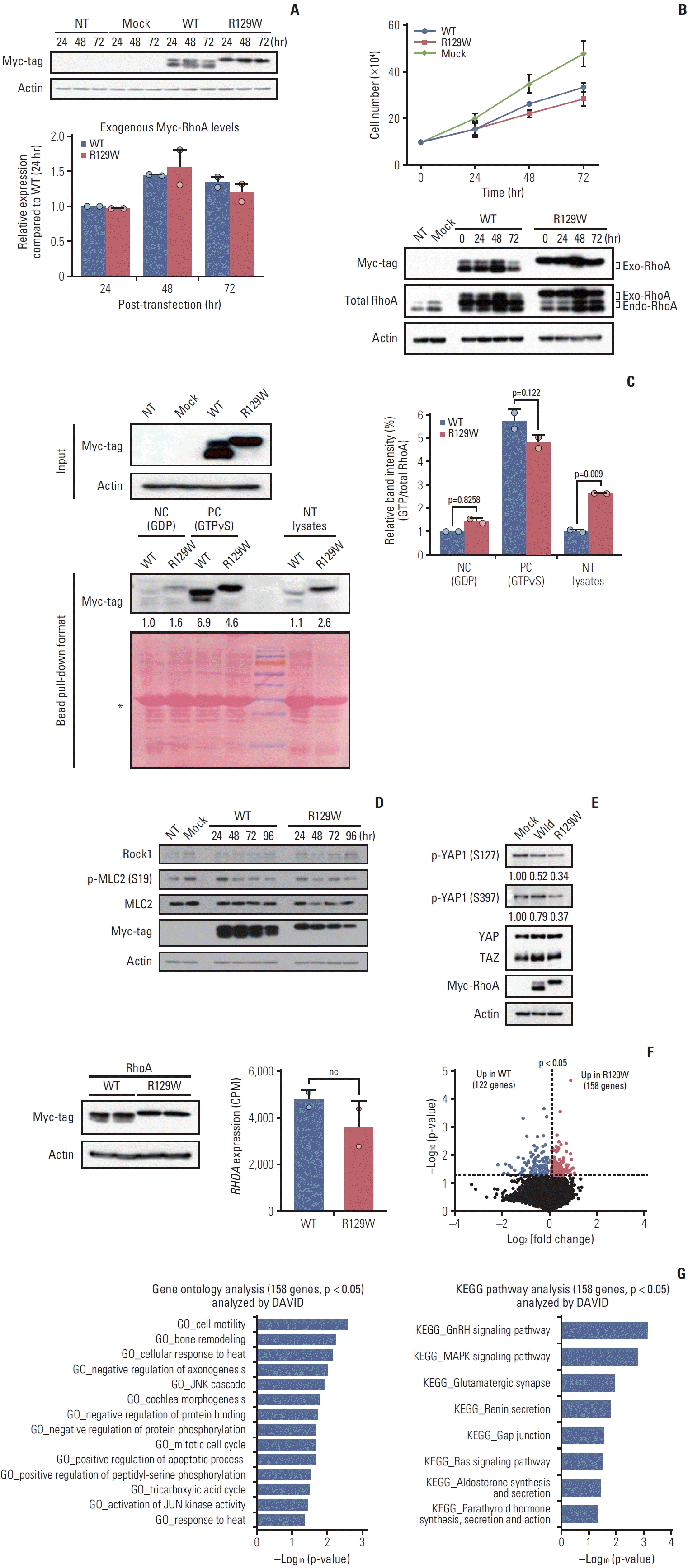
To investigate the cellular implications of the RHOA mutation (R129W) specifically independent of CDH1 mutations, we selected the MKN1, SNU216, and SNU668 gastric cancer cell lines based on genomic data analysis using cBioPortal (https://www.cbioportal.org/). These cell lines were identified to have wild-type CDH1, which excluded the effect of mutant CDH1, a well-known gene that causes gastric cancer [10,11,18,19], and wild-type RHOA genes. After performing immunoblotting analysis, the MKN1 gastric cancer cell line was selected for further experiments as it exhibited the lowest level of RhoA expression compared to the SNU216 and SNU668 cell lines (Fig. 3). Hence, we expected to observe the overexpression effect of mutant RhoA (R129W) with minimal contribution from wild-type RhoA in MKN1 cells. To generate RHOA mutation (R129W) in the Rho insert domain, a wild-type RHOA cloned plasmid (pRK5 myc-RhoA WT, #15899, Addgene) was used for site-directed mutagenesis. Next, we examined the levels of exogenous Myc-tagged wild-type RhoA (myc-RhoA) and Myc-tagged mutant RhoA (myc-RhoAR129W) by immunoblotting following transient transfection. While myc-RhoA showed two bands, myc-RhoAR129W was detected as one band, and although they have the same size (kDa), the movement of myc-RhoAR129W was confirmed to be relatively slow (Fig. 4A, upper). Under our experimental conditions, the expression levels of both proteins (mycRhoA and myc-RhoAR129W) in MKN1 cells peaked 48 hours after transfection. In addition, myc-RhoAR129W showed no significant difference in expression levels compared to myc-RhoA within 72 hours of transfection, as shown in Fig. 4A (bottom).
Based on the comparable expression levels of myc-RhoA and myc-RhoAR129W, their effects on cell growth were evaluated. However, when both proteins were overexpressed, a common phenomenon of inhibited cell growth was observed compared with the control group (mock, empty vector only). Notably, the difference in cell growth inhibition between myc-RhoA and myc-RhoAR129W proteins was not statistically significant (Fig. 4B).
Although myc-RhoAR129W is not directly involved in cell proliferation, its GTPase activity may play a potential role in regulating migration and adhesion. To further explore this, we investigated the GTP-bound (active) state of myc-RhoAR129W in MKN1 cell lysate. In the positive control [20] lysates that were incubated with GTPγS to ensure that it irreversibly occupied the GTP/GDP binding pocket and formed constitutively active forms, both myc-RhoA and myc-RhoAR129W showed similar levels of the GTP-bound state, indicating that there is no problem with the GTP-binding ability compared to the protein expression levels. Notably, in the experimental group (NT lysates), myc-RhoAR129W has over 2.5-fold (p=0.009) higher GTP-bound form than did myc-RhoA in terms of the active state (Fig. 4C). Indeed, compared with negative control lysates that were mixed with GDP to prevent the formation of new GTP-bound RhoA, the basal GTP-bound form in myc-RhoAR129W–expressing cells was slightly higher than that in myc-RhoA–expressing cells (Fig. 4C). These results indicate that RhoAR129W has a higher potential to exist in a GTP-bound state than wild-type RhoA at the cellular level.
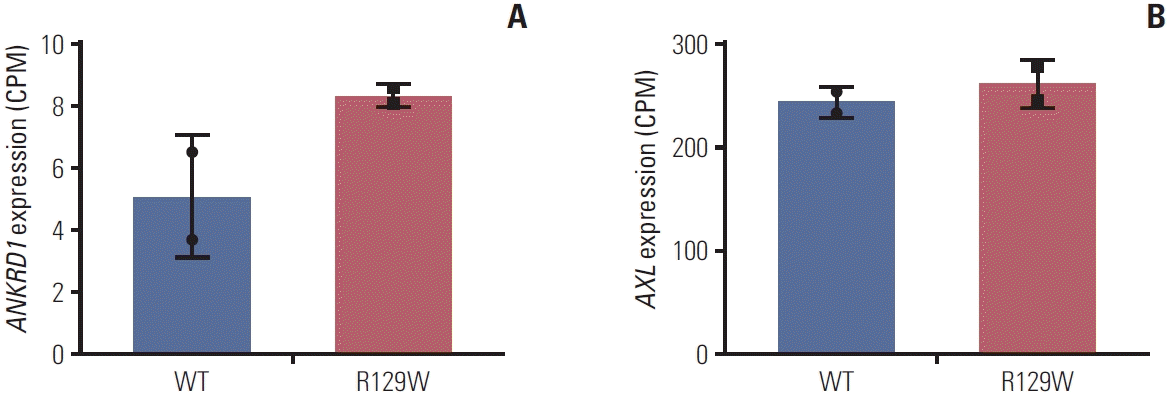
The Rho-ROCK signaling pathway is a well-known axis that regulates the reorganization of the cytoskeleton involved in cell contraction, motility, and polarity [17,21,22]. As myc-RhoAR129W–overexpressing cells showed similar cell growth to myc-RhoA–overexpressing cells, we examined ROCK1 and myosin light chain 2 (MLC2) which lies downstream of ROCK1. ROCK1-dependent phosphorylation of myosin light chain 2 (p-MLC2) is associated with stress fiber disassembly and cell detachment during migration [23]. As shown in Fig. 4D, no significant difference was observed in the expression level of ROCK1 between myc-RhoA– and myc-RhoAR129W–overexpressing cells along with no quantitative change in p-MLC2. However, the stability and activity of Yes-associated protein 1 (YAP1), which is negatively regulated by phosphorylation of Ser127/397 (p-S127/137) and regulates the extracellular matrix shape, are also known to be regulated in a Rho-ROCK–dependent manner [24]. Interestingly, p-S127/397 of YAP1 was decreased in myc-RhoAR192W–overexpressing cells (ratio, 0.34 and 0.37) compared with that in myc-RhoA–overexpressing cells (ratio, 0.52 and 0.79) compared to Mock group (ratio, 1.00 and 1.00) (Fig. 4E). We then analyzed the differential gene expression profiles of RhoAR129W versus wild-type RhoA overexpression. After confirming that the expression level of each protein was similar (Fig. 4F, left), we observed that the mRNA level (counts per million) of RhoA, which represents endogenous and ectopic mRNA, was also not significantly different after performing QuantSeq (Fig. 4F, middle). Genes that were upor down-regulated at a statistically significant (p < 0.05) level were identified. As shown in the volcano plot (Fig. 4F, right), 158 genes were upregulated in myc-RhoAR129W–overexpressing cells; conversely, 122 genes were upregulated in mycRhoA–overexpressing cells. To understand the biological significance of the158 genes upregulated by RhoAR129W, gene ontology (GO) enrichment and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analyses were performed using Database for Annotation, Visualization and Integrated Discovery (DAVID, https://david.ncifcrf.gov/). A comparison of the transcriptome of myc-RhoAR129W–overexpressing cells with that of myc-RhoA–overexpressing cells revealed that cell migration-related gene sets (e.g., ‘GO_cell motility’, ‘GO_bone remodeling’, and ‘GO_JNK cascade’), and cytoskeleton reorganization-related gene sets (e.g., ‘KEGG_MAPK signaling pathway’, ‘KEGG_Ras signaling pathway’, and ‘KEGG_Gap junction’) were significantly enriched in myc-RhoAR129W–overexpressing cells (Fig. 4G). These results suggest that RhoAR129W overexpression is related to dynamic changes in the migration/adhesion potential of MKN1 cells. In fact, ANKRD1, which encodes the transcriptional cofactor ANKRD1 protein and is upregulated in RhoAR129W-overexpressing cells (Fig. 5A), is associated with wound healing and is regulated by YAP1 [25-27]. Since both p-S127 and p-S397 of YAP1 were reduced by RhoAR129W-overexpression (Fig. 4E), it is possible that active (non-phosphorylated Ser127/397) YAP1 was involved in the increased ANKRD1 transcript levels in a RhoAR129W-dependent manner. Its involvement in these pathways highlights the possible role of RhoAR129W in epithelial remodeling during the cellular migration of cancer cells. Considering that the RhoAR129W mutation has been identified in patients with familial gastric cancer, it is crucial to gain a comprehensive understanding of its impact on gastric cancer development and the underlying mechanisms. Therefore, further studies are required to investigate whether RhoAR129W knock-in mice develop gastric cancer and to assess the therapeutic potential of targeting RhoAR129W. The tyrosine kinase membrane receptor protein AXL plays a crucial role in YAP-mediated transcriptional activity through positive feedback mechanisms [28] and acts as a mediator of YAP-dependent oncogenic functions associated with receptor tyrosine kinase inhibitor resistance, invasion, and proliferation [28-30]. However, difference between wild-type RhoA-and RhoAR129W-overexpressing cells was not significant (Fig. 5B). Taken together, these results suggest that RhoA and AXL activate YAP through separate pathways and differentially regulate YAP target genes, which requires further investigation.
We subsequently performed WES on index patients from all other families, but none of the mutations in CDH1, CTNNA1, or RHOA were detected.
Discussion
We applied next-generation sequencing technology to identify new pathogenic variants that cause HDGC and found that germline mutations in RHOA potentially contribute to the development of cancer by affecting the YAP/ TAZ pathway, one of the downstream signaling pathways of the RhoA protein. The mutation confirmed in this study, the RHOA mutation (R129W), does not belong to a hotspot, but codes for the insert domain of the RhoA protein, which can affect effector binding [17]. This suggests that RHOA may be one of the causal genes for > 60% of patients with HDGC who do not have CHD1 or CTNNA1 mutations, and may provide evidence for prophylactic total gastrectomy for family members of the patient. To our knowledge, hereditary gastric cancer caused by RHOA germline mutations has not been reported until now, and this study is the first to reveal such possibility.
According to previous reports regarding RHOA functions in gastric cancer, among the 10 cancer hallmarks published [31], mutations in the RHOA gene are known to be related to the activation of invasion and metastasis, resistance to cell death, and sustenance of proliferative signaling [32,33]. This is because the main functions of RhoA, encoded by RHOA, include cytoskeletal organization, cell adhesion, cell migration, and cell division. The RhoA protein belongs to the Rasrelated GTPase superfamily. It is characterized by switching between activation and inactivation depending on whether it binds to GDP or GTP. When bound to GTP, RhoA is activated and interacts with effector proteins to initiate downstream signaling pathways, such as the Rho/MLC and Rho/ROCK pathways, whereas when bound to GDP, RhoA is inactivated [17,34,35]. RhoA dysregulation due to RHOA mutation results in compromised tissue integrity, disrupted intercellular adhesion, increased cell motility and invasiveness, and dysregulated cell proliferation, ultimately facilitating cancer cell development, growth, invasion, and metastasis [17,32,33].
Although there was no report on the relationship between gastric cancer and germline mutations in the RHOA gene, somatic mutations in the RHOA gene in gastric cancer have been reported in several studies [32,33,35,36]. Among various subtypes of gastric cancer, RHOA mutations are primarily observed in patients with diffuse-type gastric cancer. Wang et al. [35] reported that RHOA mutation seems to be an early event because it was detected even in intramucosal diffuse-type gastric cancer. According to a study by Kakiuchi et al. [32], somatic mutations in the RHOA gene were identified in approximately 25% of diffuse-type gastric cancers, but not in a single case in the intestinal gastric cancer cohort. In general, RHOA gene mutations exist in approximately 14%-25% of diffuse gastric cancers, and the presence of RHOA mutations increases the invasiveness of the cancer [32].
Somatic mutations in RHOA are not as frequently identified as other well-known oncogenes; however, their association with many other cancers has been reported. In colorectal cancer, it is mainly noted in microsatellite-stable colorectal cancers and has been reported to be associated with mucinous adenocarcinoma. Clinically, colorectal cancers with RHOA mutations tend to be diagnosed in younger female and are mainly located in the proximal colon. An association between mutations in the RHOA gene and aggressive metastasis has been reported in breast and lung cancers, and it is also known as a susceptibility variant of angioimmunoblastic T-cell lymphoma [37]. Although research results have been reported on the effects of somatic mutations of the RHOA gene in various cancers, the association between germline mutation of the RHOA gene and hereditary cancers, such as gastric cancer, has not been sufficiently reported to date.
Recently, as many new gene analysis techniques have been introduced, opportunities for gene analysis have increased, patients have become more accustomed to genetic testing, and the cost of testing is more realistic. Concurrent with these developments, small molecule inhibitors for targeting oncogenic genes, including those for RHOA mutations, are being explored [38,39]. If a targeted therapy for RHOA germline mutations is developed, validated for safety, and widely adopted, it could provide a significantly less invasive alternative to prophylactic total gastrectomy for family members of patients with HDGC who have not developed cancer. Such an advancement could substantially improve the management of HDGC, making a significant breakthrough in the field.
Although our study has made remarkable findings, it has several limitations. First, our results suggest that the prevalence of the RHOA mutation (R129W) as a cause of HDGC may be much lower than that of the previously known CDH1 or CTNNA1 mutations. Consequently, despite the relatively high cost, genetic screening may fail to identify the causative gene, thereby providing limited guidance for future management strategies. Second, the identification of a specific mutation as the cause of HDGC does not guarantee the development of gastric cancer in all carriers because of the variability in genetic expression and penetrance among individuals. This variability can make risk assessment challenging for both individuals and their family members despite the potential benefits knowing the genetic risk factor.
In conclusion, we identified that the RHOA mutation (R129W) may serve as a potential causative gene for HDGC. However, this mutation was only identified in a single family, leaving numerous other families for which the causative gene has yet to be determined. To comprehensively understand the role of the RHOA mutation in the pathogenesis of HDGC, further is needed.
 XML Download
XML Download