

 PDF
PDF Citation
Citation Print
Print



Introduction
Many endogenous mutagenic processes or exogenous mutagens can lead to double-strand breaks (DSBs). If left unrepaired, DSBs can lead to a wide range of genomic alterations often observed in cancer. Cells employ several pathways to repair DSBs. Most prominently, homologous recombination repair (HRR) fixes DSB with high fidelity by copying the undamaged sister chromatid. In some cancers, however, HRR is dysfunctional, resulting in homologous recombination deficiency (HRD). In these cases, DSBs are instead repaired by more error-prone pathways, including nonhomologous end joining (NHEJ) and microhomology-mediated end joining (MMEJ). Among the various causes of HRD, BRCA1/2 mutations are the most frequent and well-studied. However, genomic studies in the past decade have revealed that other alterations (e.g., mutations in other genes, epigenetic silencing of target genes, structural rearrangements) could also result in the same type of genome instability as BRCA1/2 mutations, suggesting a more expansive view of HRD cases.
Research in HRD was spurred in part by the advances in therapies that exploit the underlying defect in HRR, namely poly(ADP-ribose) polymerase (PARP) inhibitors. In HRD tumors, inhibition of the PARP1 protein leads to a multitude of unrepaired single-strand breaks (SSBs), which upon replication become DSBs. PARP inhibitors also promote an increase in DSBs by trapping PARP on the DNA, forming a PARP-DNA complex which obstructs DNA replication. When cells are overwhelmed by DSBs, cell death ensues. Since the first trial demonstrated the safety and potential efficacy of PARP inhibitors in BRCA carriers in 2009, numerous studies have examined the potential to extend the benefits of PARP inhibitors beyond the BRCA-mutant tumors to other, non-BRCA-associated cancer types. Besides PARP inhibitors, platinum-based chemotherapy has also proven effective in some HRD tumors.
Given the advances in the treatment of HRD patients, accurate and cost-effective identification of patients with HRD is of paramount importance. In particular, the development of sequencing-based assays has enabled a more precise, genome-wide characterization of genomic instability that accompanies HRD. With the increasing accessibility of genome sequencing methods in clinical settings, the design and use of optimal assays is likely to result in more effective trials and an expanded patient population that will benefit from PARP inhibitors.
In this review, we focus on genome-based methods for identifying patients with a defective HRR pathway, resulting in HRD. After briefly reviewing key components of the HRR pathway, we describe the genomic consequences of HRD, especially copy number variations (CNVs) and structural variations (SVs) observed in whole-genome sequencing (WGS) data. Then, we describe current sequencing platforms and recent advances that have increased the accuracy of HRD detection, which often involve combining multiple genomic features of HRD into a statistical framework. We pay particular attention to the use of ‘mutational signatures,’ which aim to capture a specific set of genomic features associated with distinct mutagenic sources. The last section highlights the clinical application of HRD detection, including a review of platforms used in key PARP inhibitor trials.
Homologus Recombination Deficiency
1. HRR pathway
The HRR pathway starts with the recognition of DSBs by the MRN complex (MRE11, RAD50, and NSB1) and subsequent activation of ATM, which leads to further activations of BRCA1, BRCA2, and PALB2 [1]. The exonuclease component of the MRN complex (in the presence of functional BRCA1) leads to DNA end resection from the 5′ to the 3′ end, generating a 3′ overhang coated by replication protein A (RPA). Then, RAD51 together with BRCA2 replaces RPA, assisted by BRCA1 and PALB2 [2]. This RAD51-DNA nucleoprotein complex initiates D-loop formation and strand invasion. Through such intricate interplay of different steps of repair mechanisms, the cell ensures accurate replication and maintains genome stability.
During the cell cycle, HRR preferentially occurs during the S/G2 phase when end resection can be readily performed, as end-protecting factors (53BP1 and REV7) are inactivated by CDKs [3,4]. In contrast, during other phases of the cell cycle or when HRR is defective, there is a shift towards non-HRR mediated, error-prone DNA repair, namely NHEJ and MMEJ. The NHEJ process, mediated by the Ku70-80 heterodimer complex and DNA-PKc, directly ligates the two broken ends, often creating characteristic mutations, deletions, or inter-chromosomal translocations [5]. MMEJ is mediated by PARP1, which regulates the balance between NHEJ and MMEJ through its competition with the Ku70-80 complex. Following the DNA resection step, the MMEJ process utilizes polymerase theta and microhomology features (homologous DNA sequences of 5 to 25 bp) at DNA breakpoints to join the broken ends [6]. More details on these mechanisms are reviewed elsewhere [7].
2. Characteristic footprints of genomic instability in HRD
An intact HRR pathway is crucial for genome integrity and tumor suppression, and its loss is a prevalent cause of cancer in tumor types such as ovarian cancer and triple-negative breast cancer. The leading cause of HRD is the bi-allelic loss of BRCA1 or BRCA2, most commonly due to mutations on one allele and loss of heterozygosity (LOH) of the other allele, and promoter hypermethylation of BRCA1 [8]. Beyond BRCA1/2, loss of other central HRR genes can also lead to HRD [9]. In addition to their DSB repair function, BRCA1/2 are involved in other key cellular processes such as maintenance of replication fork stability, R-loop resolution, and protection of telomere integrity, all of which contribute to maintaining genome integrity [10,11].
With impaired HRR, the increased contribution of NHEJ/MMEJ leads to characteristic patterns of genomic instability (some refer to these as “genomic scars”). Comparison of HRD and non-HRD (i.e., homologous recombination proficient, HRP) cell lines and tumors have revealed several features of HRD-specific genomic instability involving LOH, allelic imbalance, and chromosomal rearrangements [8,9,12,13]. These features all reflect improper repair of DSBs and are linked to each other—allelic imbalance, for instance, can be caused by copy number—neutral LOH, deletion, or monoallelic copy number gain. In one study, the authors examined different summary statistics of chromosomal alterations and found the number of chromosomal regions with telomeric allelic imbalance (TAI)—allelic imbalance that extends to the telomeric end of a chromosome—to be the most accurate predictor of sensitivity to cisplatin in breast cancer cell lines and associated with impaired HRR [12]. In another study, the authors found frequent large-scale chromosomal rearrangements to be characteristic of BRCA1-associated genomic instability in breast cancers and proposed the number of large-scale state transitions (LST)—chromosomal breaks between adjacent regions of at least 10 Mb—as a robust predictor of BRCA1 status [13]. A commercial HRD test used in many key clinical trials defined the genome instability score (GIS) as a simple sum of LOH events (defined as the number of sub-chromosomal LOH regions > 15 Mb), TAI, and LST scores (see later sections for more details) [14]. These measures were established more than a decade ago based on single nucleotide polymorphism (SNP) arrays, prior to the widespread use of WGS. Therefore, although they attempted to capture genome-wide characteristics of genome instability, the detected features were of low resolution, not allowing inference of detailed rearrangement types.
3. Clinical significance of HRD
Accurate detection of HRD in cancer patients is important because of its therapeutic implications. An efficacious mode of intervention is the PARP inhibitor, which targets PARP1 and PARP2 enzymes. PARP inhibitors restrict the base excision repair pathway for repairing DNA SSBs, leading to an accumulation of unrepaired SSBs, which is then converted into DSBs during replication [15]. Since cells with BRCA1/2 mutations or HRD phenotypes cannot efficiently repair DSBs, PARP inhibitors lead to synthetic lethality. Moreover, PARP inhibitors cause PARP1/2 trapping, which could be associated with drug potency among different types of PARP inhibitors [16]. The first human trial using PARP inhibitors was conducted in 2009, in which 60 patients with multiple cancer types received olaparib in recurrent settings [17]. Responses were observed in breast, ovarian, prostate, and pancreatic cancers, which are BRCA-associated cancers. Since then, PARP inhibitors have become standard care in ovarian cancer. Most notably, in the SOLO-1 trial, olaparib for front-line maintenance in ovarian cancer patients with BRCA mutations was associated with a progression-free survival (PFS) benefit of 36 months compared to placebo, with a hazard ratio of 0.30 (95% confidence interval, 0.23 to 0.41) [18]. A recently published 7-year follow-up suggests that patients continued to derive benefits even after 2 years on olaparib [19]. Although not as dramatic, efficacy of PARP inhibitor was also shown in metastatic or recurrent settings and in BRCA-associated cancer types other than ovarian, as several trials have highlighted [20-22]. Importantly, studies have suggested that patients with HRD disproportionately benefit from PARP inhibitor even when they are wild-type for BRCA (see later sections for more details). Therefore, an accurate HRD detection strategy can be pivotal for refining patient selection criteria and the expansion of cancer types for which PARP inhibitor is not currently considered.
Mutational Signature Analysis
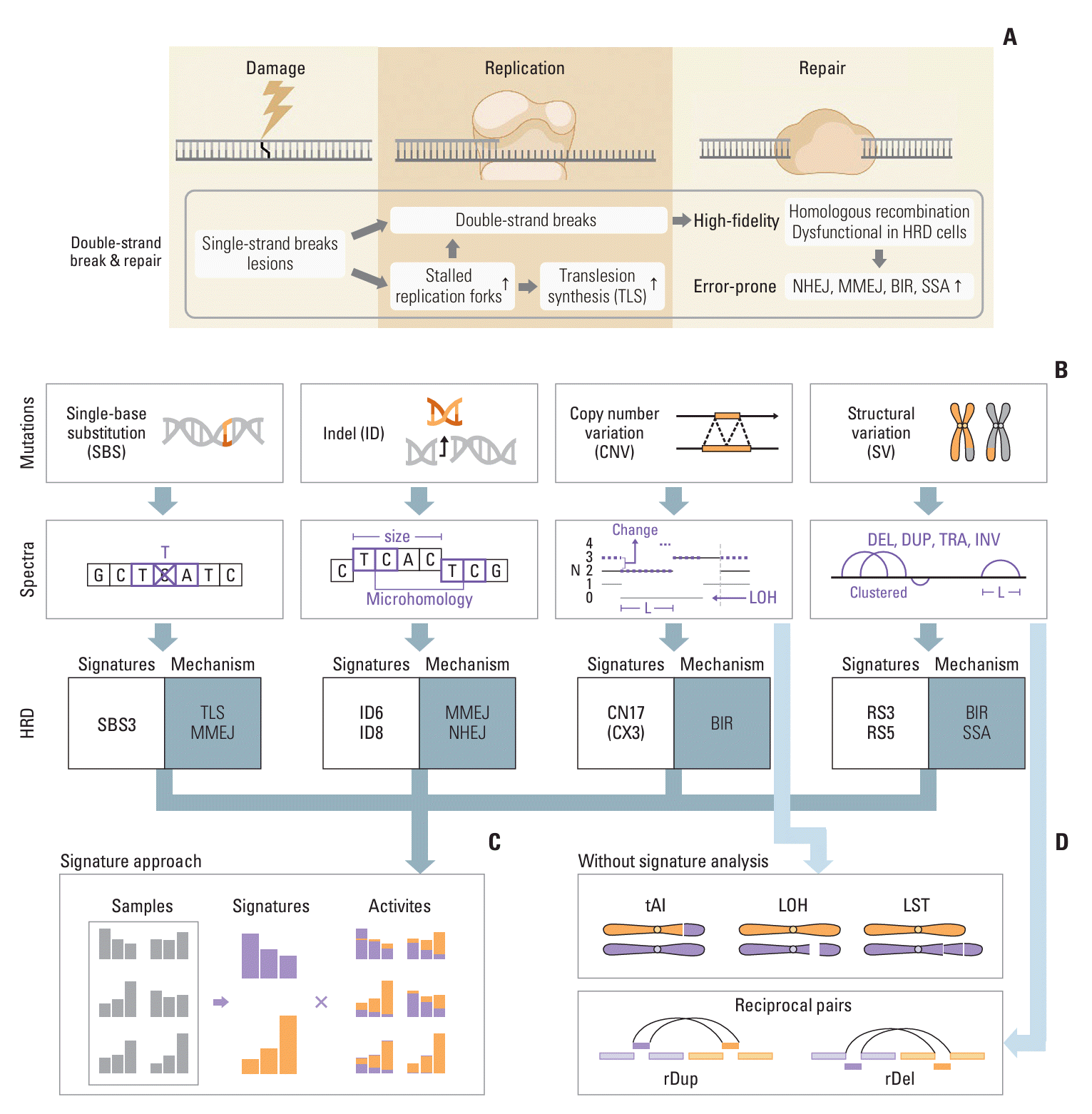
To understand the advances in HRD prediction algorithms, it is necessary to understand the fundamentals of ‘mutational signatures,’ an analytical framework first described a decade ago [23] and now routine in cancer genome analysis. Here, we describe the key concepts and basic properties of mutational signature analysis in non-mathematical language, with a focus on HRD-associated signatures. A schematic summary of signature analysis in the context of HRD is shown in Fig. 1. More detailed reviews of mutational signature analysis are available elsewhere [24-26].
1. Basic concepts and single base substitutions
Signature analysis aims to decipher the etiology of specific mutational processes based on their characteristic patterns in the genome. In the simplest case of point mutations, different types of single base substitutions (SBSs) are enriched in cancers of different tissues. For example, lung and liver tissues are specifically subject to DNA damage induced by tobacco smoking, resulting in an enrichment of C>A substitutions in the associated tumors. Furthermore, the DNA sequence context surrounding the substituted bases often provides additional information on the underlying mutagenic process. For instance, spontaneous deamination of 5-methylcytosine—an endogenous mutational process operative in nearly all tissues—creates C>T mutations almost exclusively in the CpG context. Standard signature analysis for point mutations thus centers on mutation frequency spectra of trinucleotides, consisting of the substituted base together with one adjacent base on each side, yielding 96 combinations given the four bases. It is possible to extend to larger contexts, e.g., two bases on each side for a pentamer, but such analyses appear to yield limited additional insights, at least with currently available data.
As multiple mutagenic processes are typically active in a cancer genome, its mutational spectrum reflects the cumulative effect of such processes over time. To identify what mutagenic processes might be present (de novo signature discovery), a standard approach is to perform non-negative matrix factorization (NMF) on the mutational type-by-sample matrix. Similar to principal component analysis (PCA), NMF aims to identify the building blocks (i.e., signatures) that best summarize the data; the difference compared to PCA is that all elements of the signature and the coefficients used in decomposition must be positive.
By applying NMF to large numbers of cancer genomes, catalogs of reference mutational signatures have been derived. One popular catalog is the COSMIC Mutational Signatures, which has been an indispensable resource in the field. The latest version (v3.4, 2023) was largely derived from 2,780 cancer genomes. An additional > 1,800 genomes and > 19,000 exomes were used to assess stability and reproducibility [23,27]. Several curated signatures derived from other sources were also included in the catalog. For SBSs, the COSMIC catalog contains 86 signatures, 67 of which are thought to be of biological origin, whereas the rest are suspected to be possible sequencing artifacts. Such biological annotations are possible via population association studies or experimental studies, e.g., by the sequencing of cell lines after exposure to mutagens or the knock-down of genes with known function in DNA repair. Examples of signatures with well-established etiologies include SBS1 (spontaneous deamination of 5-methylcytosine), SBS2/SBS13 (activity of the APOBEC cytidine deaminases), SBS4 (tobacco smoking), and SBS7 (UV light exposure).
Catalogs of reference mutational signatures are evolving. As more genomes of cancer and other diseases are being sequenced, new signatures will be discovered [28]. Known signatures may also be refined or revised. For example, with more samples, there may be sufficient statistical power to further separate an existing signature into multiple ones [29]. It is important to note that deriving reference catalogs is a challenging process that can be error-prone and often requires manual intervention. For example, one needs to be cautious when analyzing cohorts with hypermutated samples, as a few hypermutated samples may bias the signature discovery process by contributing a disproportionately large number of mutations. Particularly challenging are ‘flat’ signatures—where the spectra are more or less even across the triplets—as the underlying signatures become difficult to separate mathematically. Therefore, it is important to verify the robustness of discovered signatures in orthogonal datasets or through experiments.
Given that a fairly stable catalog has been generated, a common procedure in signature analysis is to ‘refit’ a given spectrum with a combination of catalog signatures, i.e., estimating the weighting factor for each signature so that the signatures sum to the original spectrum with minimum error. As is the case for deriving reference catalogs, refitting is not a clearly-defined process. Depending on the choice of the algorithm and the thresholds therein, a sample may be found to contain a range of different signatures. To decrease the likelihood of a false positive signature, one should consider limiting the analysis only to a subset of signatures that are likely to be present in a given tissue as informed by previous studies of large cohorts, although this will prevent the discovery of a new biological process operative in the sample.
2. Alterations beyond SBSs
Recent signature analysis has incorporated mutation types beyond SBSs, including double-base substitutions (e.g., CC>TT), indels, CNVs, and SVs. These alterations can be identified from sequencing data using a wide range of computational tools, as summarized in our recent review [30]. Double-base substitutions are relatively rare, with a burden of about 1% of that of SBSs, and their signatures are defined without the surrounding sequence context to avoid the extremely large number of possible categories. For other mutation types, ad hoc definitions are used to strike a balance between capturing sufficient details of the mutation context and avoiding an excess of categories, which would lead to overly sparse spectra. Specifically, for indels, it is not feasible to enumerate all possible inserted or deleted sequences. The standard indel signature definition thus classifies indels primarily based on their sizes and whether they occur at repeat regions (except for 1-bp indels, which are further separated based on the affected nucleotide). Deletions with microhomologies are further put into separate classes, as they are often informative of specific mutational processes [27,31]. Signature definitions of CNVs and SVs are even more challenging and less established [32-36]. The current COSMIC CNV signatures are mainly defined by the total copy number (1, 2, 3-4, 5-8, 9+) and the size (1-100 kb, 100 kb-1 Mb, 1-10 Mb, 10-40 Mb, > 40 Mb) of each segment, while incorporating the heterozygosity status.
3. Mutational signatures associated with HRD
Previous studies have linked HRD with specific mutational signatures, most notably SBS signature 3 (SBS3), characterized by a roughly uniform distribution across all 96 substitution types. First identified as correlating with BRCA1/2 mutations in breast cancers [36,37], SBS3 was later found to be associated with other HRR gene alterations as well, such as epigenetic inactivation of BRCA1 and RAD51C or PALB2 mutations [38]. The association between SBS3 and HRD was validated by knockout experiments of BRCA1/2 genes initially [39] and other HRR-genes later [40]. The mechanistic origin of SBS3 in HRD tumors is still an open question. Proposed mechanisms involve polymerase theta in MMEJ [41] or REV1/REV3L in translesion synthesis [42], although in another study the double knockout of BRCA1 and REV1 still exhibited SBS3 [43].
Other non-SBS signatures associated with HRD are indel signatures ID6 and ID8, as well as rearrangement signatures RS3 and RS5 [27,36,38-40]. ID6 mainly consists of ≥ 5-bp deletions with overlapping microhomologies at deletion boundaries, suggesting an MMEJ origin. ID8 also contains ≥ 5-bp deletions, but does not show an enrichment of long microhomologies, suggesting NHEJ as its underlying cause. In addition, ID8 was found to be enriched in tumors with topoisomerase mutations [27,44]. Rearrangement signatures are defined based on the type (deletions, tandem duplications, inversions, and translocations) and size of the rearrangements, as well as whether they occur in clusters [36]. Clustered rearrangements are likely to have originated from the same complex event, thus pointing towards distinct mutational processes. The two HRD-associated rearrangement signatures correspond to different underlying causes of HRD. Specifically, RS3, characterized by small tandem duplications of < 10 kb is associated with BRCA1 mutations, whereas RS5, characterized by deletions of 10-100 kb is associated with BRCA2 mutations [45] and enriched in cases with PALB2 or RAD51C losses [40,46]. Of note, RS5 is also present at lower amplitudes in HRP tumors, such as a large fraction of lung, liver, and cervical cancers and melanomas [47]. Furthermore, enrichment in deletions with homeology (approximately homologous sequences of > 50 bp) in BRCA2 mutated samples suggests a potential link between single-strand annealing and RS5 [48,49]. Finally, a recent study using “linked-read” sequencing (Illumina technology that can track fragments from the same > 50 kb DNA molecule but is now discontinued due to patent issues) found complex SVs in BRCA1/2-deficient tumors that were missed by short-read data. Rather than balanced translocation or inversion, near-reciprocal rearrangement with copy loss or gain of the intervening regions were found to be enriched in these tumors [49].
Although a COSMIC mutational signature catalog has also been generated for CNVs, it has not been as informative for HRD prediction. First, other features, especially microhomology-mediated deletions and SBS3, are more effective than a CNV-based measure. Second, GIS score or its variants (e.g., HRD index [50]) already capture the key features in large CNVs, even though those scores are ad hoc measures of genome instability. In two recent CNV signature analysis approaches [32,34], the copy number patterns are defined using copy number states, heterozygosity, segment length distributions, breakpoint density, and other related measures. Not surprisingly, the signatures linked with HRD in both papers, CN17 [32] and CX3 [34], have segment length distribution similar to that used to define components of GIS (each paper used its own CNV catalog with different signature names). Third, CNV signatures are more complicated to construct due to the difficulty in identifying whole-genome duplication regions accurately. For example, the most common copy number states in CN17 are 3 and 4, suggesting that this signature may be capturing events only in samples with whole-genome duplication rather than a broad signature that applies to all HRD samples.
4. Future improvements in mutational signature analysis
Signature analysis has become a standard tool in interpretation of somatic mutations, but there are several shortcomings with current approaches. For example, the observed mutations and their signatures reflect the residual imprints of errors after the repair processes have taken place, rather than the initial DNA damage. This becomes apparent when DNA damage introduced in cells with a deficient repair pathway leads to specific mutational patterns [51,52]. To address this problem, explicit modeling of the damage-repair interaction has been proposed [53]. Another challenge is that mutations do not accumulate uniformly along the genome, as both DNA damage and repair vary depending on locusspecific features such as transcription activity, replication timing, presence of DNA-binding proteins, and epigenetic modifications [52,54]. New signature analysis methods are being developed to incorporate these additional attributes [55]. Even within the standard signature analysis framework, important methodological issues remain. In particular, the NMF algorithm at the center of de novo signature discovery methods has an intrinsic problem of producing non-unique solutions; the procedure of deriving a reference signature catalog needs a method less dependent on manual interventions; and signature refitting is lacking a statistically sound algorithm that minimizes incorrect signature assignments. We have recently developed Mutational Signature Calculator (MuSiCal), a rigorous analytical framework for signature analysis that incorporates several algorithms to tackle some of these issues [56].
Current Assays for HRD Detection
Existing HRD detection approaches can be classified based on what genomic information they rely on and which sequencing platform they use. A summary of genomic information relevant to HRD detection for each sequencing platform is shown in Table 1 [50,57-59].
1. Panel-based HRD detection method
An early approach for HRD detection, prior to the development of mutational signatures, is based on mutations of genes associated with HRR. These typically include PALB2, BARD1, BRIP1, RAD51B, RAD51C, RAD51D, ATM, FAAP20, CHEK2, FAN1, FANCE, FANCM, and POLQ among others [60]. However, applying this gene-based approach to clinical HRD detection has been challenging for several reasons. First, clinical studies have utilized slightly different sets of HRR genes, and the low prevalence of mutations in individual HRR genes makes it difficult to assess the clinical significance of each gene. Second, mutations of HRR genes, when followed by an LOH event (thus creating a bi-allelic loss), are more likely to be pathogenic. But determining the monoallelic vs. bi-allelic status of a mutation is difficult, especially when the tumor purity is low. Third, evaluating the pathogenicity of missense mutations in HRR genes is challenging, as variants of unknown significance are common except for the well-studied BRCA1/2.
Clinical trials involving PARP inhibitors have frequently utilized panel sequencing to identify patients with BRCA mutations or other HRR gene mutations in prospective randomized trials. Their inclusion criteria were often shaped by the initial trials that led to the approval of PARP inhibitors in each cancer type. Regardless of cancer type, BRCA mutations were associated with a favorable response to PARP inhibitor, whereas a clinical benefit was not consistently observed for non-BRCA, HRR mutations. In prostate cancer, BRCA1, BRCA2, ATM, and other 12 prespecified HRR genes were used as inclusion criteria for the first trial that led to PARP inhibitor approval in prostate cancer [61]; thus, non-BRCA, HRR genes are still frequently used as the inclusion criteria. In contrast, in ovarian cancer, post-hoc analyses of major PARP inhibitor trials have shown mixed findings—either a trend of improved response or no improvement in HRR-mutated subgroup [62,63]. There is ongoing research to understand the clinical significance of non-BRCA HRR gene mutations, such as identifying the tumor-specific context in which these mutations arise and determining which specific mutations drive therapeutic response.
Although panel sequencing is routinely performed in many cancer hospitals to detect cancer-associated hotspot mutations, the number of identified mutations from panels is generally > 1,000-fold smaller than from WGS, rendering standard mutational signature analysis approaches ineffective. The HRD-associated SBS3 is particularly difficult to detect because its “flat” profile requires more mutations to ascertain its presence with confidence. To ameliorate this situation, we have previously developed a computational method called Signature Multivariate Analysis (SigMA) to identify SBS3 from targeted gene panels [57]. The key innovations were leveraging existing WGS data to learn expected signature combinations for each tumor type and developing a robust classification framework with machine learning. We validated our method by comparing SigMA-derived SBS3 calls with BRCA mutations and comparing results obtained from paired panel and exome data from the same patients [64]. We also showed that the SBS3 status inferred from panel data using SigMA predicted the response of breast cancers to PARP inhibitors and outperformed existing clinical HRD classification strategies [64,65]. Despite the reduced sensitivity and specificity compared to WGS, SigMA enables mutational signature analysis to be performed using panel sequencing data.
2. Detecting HRD-associated genome instability based on copy number profiling
Another popular approach for HRD detection is to measure HRD-associated large-scale copy number alterations, ideally in conjunction with point mutation detection in HRR genes. The commercial test Myriad myChoice provides a ‘Genome Instability Score’ (GIS) based on the three copy numberbased statistics described earlier (TAI, telomeric allelic imbalance; LOH, loss of heterozygosity; and LST, large-scale state transitions), along with the BRCA1/2 mutation status. A GIS cutoff of ≥ 42 was defined based on 95% sensitivity to detect breast cancers with BRCA1/2 mutations and BRCA1 promoter methylations [14]. GIS was initially computed from SNP arrays, but now high-throughput sequencing is used. The Foundation Medicine assay FoundationOne uses the BRCA1/2 mutation status and the genomic LOH percentage based on targeted panel sequencing to detect HRD. In addition to HRD status, it reports pathogenic mutation status of 324 cancer-related genes, including HRR-associated genes.
3. Enhanced HRD characterization with WGS
WGS enables the most unbiased and comprehensive quantification of mutational signature. In addition to a larger number of single nucleotide mutations, WGS allows detection of various structural changes, such as large insertions, deletions, and rearrangements [30], thus enabling analysis of copy number and rearrangement signatures. Although it is possible to detect large-scale CNVs from exome data, WGS enables more accurate CNV analysis, with the resolution of CNV dependent on sequencing coverage.
The first method for HRD detection based on WGS is HRDetect (2017), which uses mutational signatures SBS3, SBS8, RS3, and RS5, deletions with microhomology, and the HRD index (a variant of GIS) as covariates for lasso logistic regression modeling. HRDetect was trained on breast cancer samples with BRCA1/2 deficiency, and this classifier was later also applied to other tumor types [47,50]. The most impactful predictors were the proportion of deletions with microhomology and SBS3, followed by RS3, RS5, and HRD index. Another WGS-based algorithm is CHORD (Classifier of HOmologous Recombination Deficiency) [58], which utilizes a random-forest classifier with 29 features, many of which correspond to components of the mutational signatures used in HRDetect. The two features that contribute the most in CHORD were ≥ 2 bp deletions with flanking microhomology and duplications of size 1-100 kb.
These two methods when applied to multiple tumor types provided largely concordant results [58], e.g., having the deletions with ≥ 2 bp flanking microhomology as one of the most predictive features of HRD. Pan-cancer HRD classification is a challenging task. For HRDetect, training based on breast cancer data may not provide the optimal classification performance for other tumor types. For CHORD, although training was performed in a pan-cancer setting, the positive training set was largely dominated by tumor types typically associated with HRD, such as breast and ovarian cancers. To resolve this problem, more WGS samples must be available publicly for tumor types with low HRD prevalence, so that tumor type-specific training can be performed.
A major question in the design of a WGS assay for HRD is the sequencing coverage. If the aim is simply to measure genome instability alone, it is possible to obtain reasonably accurate calls from shallow coverage. For example, ShallowHRD used ~1× data to measure large-scale CNVs by a statistic similar to LST for SNP arrays, and found a high-concordance with the SNP array-based calls [59]. The same group subsequently showed that shallowHRDv2 had 94% agreement with Myriad myChoice across 449 high-grade ovarian cancers and that patients with HRD according to shallowHRDv2 had longer PFS [66]. While we suspect that not having other features (e.g., microhomology-mediated indels and SBS3) make this less sensitive and less precise, shallow coverage sequencing may be a simple, low-cost alternative to currently available commercial tests.
4. HRD testing in PARP inhibitor trials
In this section, we summarize the key clinical trials that led to the Food and Drug Administration (FDA) approval of PARP inhibitors, focusing on HRD testing platforms and the efficacy of PARP inhibitors with respect to HRD stratification (Table 2) [18,20,61,67-78]. Across various trials and cancer types, PARP inhibitor efficacy varies widely, likely due to differences in cancer specificity and the treatment setting, since PARP inhibitors had to be incorporated into existing treatment flow. However, consistently across trials, patients with pathogenic BRCA mutations disproportionately benefit from PARP inhibitors compared to those with BRCA wild-type disease. Consequently, PARP inhibitors have been approved for patients with BRCA mutations in ovarian, breast, prostate, and pancreatic cancers. Beyond BRCA mutations, patients with HRD tumors also benefit from PARP inhibitors compared to those with HRP tumors. However, this summary also highlights the considerable differences in how HRD was tested and defined across these trials (e.g., HRR mutation or GIS, SNP array or sequencing, commercial test or in-house assay); the approved indications are driven by the diagnostic platforms available at the time of the study. Efforts to further refine HRD classification and expand the indication for PARP inhibitors to other cancer types would require an understanding of the biological differences in cancer types that underlie the HRD phenotype, as well as careful design of prospective trials with respect to inclusion criteria, testing platforms, prespecified analysis plans, and sample size.
Perspectives for the Future
1. Incorporating WGS-based HRD detection in the clinic
In addition to more comprehensive HRD detection, WGS provides a wealth of information about the genome-wide mutational profile of the tumor, thereby enabling other types of analysis that should be of interest to physician-scientists. Despite these advantages, WGS is not typically incorporated in large prospective trials or in clinics yet. To implement WGS-based assays, several issues must be resolved.
With the introduction of the latest Illumina sequencers (e.g., NovaSeq X) in 2023, standard 30× short-read WGS for germline analysis is now < $400, including the cost of library preparation. If HRD detection is the sole purpose, low coverage WGS can be performed at a much lower cost. For research projects, tumors are generally sequenced at a higher depth (e.g., 60-90×) for somatic variant analysis, which amounts to ~$1,000. With this reduction in sequencing cost, even the cost of high-coverage WGS is relatively small compared to the current cost of FDA-approved HRD tests ($3,500 for Foundation CDx or Foundation liquid CDx; $4,000-6,000 for Myriad myChoice CDx). Thus, sequencing cost is no longer the main bottleneck. Furthermore, several new short-read platforms (Ultima Genomics, Complete Genomics, MGI, Element Biosciences, Singular Genomics) have been introduced just in the past couple of years, with many of them advertising $200 genomes. The cost reduction in long-read sequencing on the PacBio and Nanopore platforms is also changing the landscape of WGS studies.
A much greater challenge for WGS than the sequencing cost is the expertise and infrastructure for downstream analysis. At > 100 GB per 30× genome, any sequencing effort will require processing and storing of terabyte-scale data, requiring a substantial investment in a team of bioinformatics experts and infrastructure [79]. Comprehensive variant identification and interpretation for WGS is an art rather than a science, especially for non-coding variants, structural variants, and coding variants of unknown pathogenicity. Therefore, scientific leadership as well as recruitment and retention of experienced bioinformaticians will be needed. For infrastructure, an existing institutional computing environment may be sufficient for small projects, but additional servers are likely to be needed for larger ones. It is possible to avoid building a local computing environment by utilizing a cloud-based platform such as Amazon Web Services (AWS) or Google Cloud, but this necessitates additional level of informatics expertise or subscription to an informatics platform by a commercial vendor. Computing and storing data on the cloud can also be costly and may require regulatory approval in some institutions and/or countries.
Despite these limitations, WGS studies are continuing to grow in scale and are discovering new insights on driver mutations [80]. For instance, a recent study [81] from the UK 100,000 Genomes Project analyzed > 13,000 solid tumors with treatment outcome data, finding that some tumor types showed actionable structural variants in more than 10% of the cases. Importantly, that study also found that HRD was identified in 40% of high-grade serous ovarian cases with 30% linked to pathogenic germline variants.
Regardless of the progress on WGS data generation and analysis, panel sequencing is likely to continue in the clinic due to the extremely high-coverage needed in that setting. Gene panels in a real patient setting are often sequenced at > 1,000× to detect clinically-relevant variants with very low variant allele fraction (VAF). A study of > 5,000 clinical samples in Samsung Medical Center showed that 10%-25% of hotspot mutations have VAF under 5% and that a substantial fraction of these might be missed if the coverage is lower. For example, 24% of EGFR T790M had < 5% VAF, and 25% of all T790M mutations would be missed if the sequencing coverage is 100× rather than 1,500× [82]. These VAFs are low partly because tumor purity of clinical samples is often low, whereas VAFs in research sequencing projects tend to be higher because a tumor purity threshold is often imposed, e.g., the distribution of VAFs in The Cancer Genome Atlas data are substantially higher than that of real clinical samples [82]. Performing WGS at 1,000× is impractical not only because of the cost, but because of the difficulty in handling and analyzing such data, as one sample would generate more than > 10 TB of data and many WGS algorithms would fail to run. One likely scenario in a research hospital setting would be to perform both a high-coverage panel for hotspot mutations and ~60-90× WGS for a comprehensive characterization of mutations.
Another challenge for WGS adoption is the question of whether high-quality data can be obtained from formalin-fixed paraffin-embedded (FFPE) samples. Formalin-induced DNA damage in FFPE samples results in artifact mutations, which can confound the identification of true variants. In some cases, the problem of artifactual point mutations, mostly C to T transitions due to deamination of cytosine residues, can be ameliorated with a series of filters. For mutational signature analysis, FFPE mutations correspond to SBS30 and can thus be isolated. FFPE samples also contain a large number of artifactual chimeric reads [83] that can be mistaken for structural alterations. One simple way to lessen the impact of FFPE-specific artifacts is to increase coverage, as artifacts are less likely to occur at the same position repeatedly and higher coverage makes it easier to distinguish them from true mutations.
2. Analyzing PARP inhibitor resistance in longitudinally collected samples
A topic of growing interest is the mechanisms of acquired PARP inhibitor resistance such as BRCA reversion mutations. These mutations refer to the secondary mutations that can restore the native reading frame of the mutated gene, thereby reducing sensitivity to platinum or PARP inhibitors. One meta-analysis examined BRCA reversion in four BRCA-associated cancer types (breast, ovarian, prostate, and pancreatic, with the majority being ovarian) after platinum or PARP inhibitor treatments and found reversion mutations in 22% of BRCA1 and 30.7% of BRCA2 cases [84]. BRCA reversion rates can vary substantially depending on prior therapy and response to prior therapy, e.g., one study (n=69) reporting BRCA reversion rates of 2% in platinum-sensitive, 13% in platinum-resistant, and 18% in platinum-refractory cancers [85]. In terms of the mechanisms responsible for BRCA reversion, the majority of the reversion mutations were deletions mediated by NHEJ, often involving sequence microhomologies indicative of MMEJ, especially for BRCA2 [84,86]. However, other reversions do not seem to utilize microhomologies, suggesting that further research is needed to understand the DNA repair or mutagenic processes that generate the reversion mutations. There are also non-BRCA associated PARP inhibitor resistance mechanisms that are either homologous recombination-dependent (i.e., the change from HRD to HRP phenotype) or independent, as detailed in separate reviews [87-89].
For a refined view of how PARP inhibitor resistance evolves under therapeutic pressure, an ideal strategy would be to non-invasively and serially collect circulating tumor DNA (ctDNA) with liquid biopsy. Recent studies showed that resistance mechanisms to PARP inhibitor can be detected from plasma [86,90], with one study finding BRCA reversion mutations in ctDNA in 60% of patients who develop resistance and that detection of a reversion mutation was associated with shorter time to progression [86]. To bring these scientific findings into clinic, a prospective clinical trial is needed address how these specific resistance mechanisms may inform clinical decision making. Such a trial would require meticulous genomic characterization of the tumor at initial diagnosis and at the time of PARP inhibitor resistance acquisition, accompanied by genomic sequencing of serially obtained blood samples.
In addition to liquid biopsy, another emerging technology relevant for HRD studies is single cell RNA sequencing (scRNA-seq). Transcriptomic analysis using scRNA-seq will help characterize the heterogeneity of the cell populations that display HRD signatures, including the interaction between DNA repair-deficient cells and their tumor microenvironment [91]. Recent advances in computational methods allow identification of point mutations and copy number profiles from scRNA-seq data [92,93], which can potentially be used to identify single cells directly from their mutational profile rather than relying on their gene expression patterns. Applications of recent methods in single cell multi-omics [94], e.g., sequencing DNA and RNA simultaneously from the same single cells, may also prove informative in characterizing genetic and phenotypic heterogeneity of HRD samples.
Conclusion
The methods for detecting HRD continue to evolve as we obtain a more accurate view of the mutations associated with HRD. In particular, WGS is allowing us to characterize HRD-associated genomic instability more accurately and thus refine the GIS currently being used in HRD assays. Although incorporation of WGS in clinical trials requires additional costs and bioinformatics expertise, such data will provide more accurate prediction of HRD status as well as better assessment of the correlation between HRD status and response to therapy. Both for panel and WGS data, mutational signature analysis has provided a more robust approach to identify HRD, and this approach should also be applicable to liquid biopsies, which will be particularly useful for investigating acquired resistance mechanisms.
Despite the tremendous progress in expanding the potential pool of patients for PARP inhibitor treatment, the eligibility criteria and treatment settings (early vs. late stage, front-line vs. recurrent) are highly variable across tumor types. A more systematic understanding of de novo and acquired resistance, tumor type-specific effects, impact of front-line therapy, and the correlation between genotype and genomic instability will be needed to design more effective therapeutic strategies for patients with HRD.
 XML Download
XML Download