

 PDF
PDF Citation
Citation Print
Print



Introduction
Breast cancer is the most common cancer among women and is increasing globally, including in Korea [1-3]. Recent research has led to significant advances in new therapeutics and precision medicine, and active clinical applications of the novel approaches resulted in improved prognosis of patients with breast cancer [4-6].
Despite these advances, triple-negative breast cancer (TNBC) remains the most challenging subtype due to its aggressive and high metastatic potential even after a good response to standard systemic chemotherapy [7]. Since the first classification of TNBCs to six molecular subtypes based on mRNA expression, significant efforts have been made to elucidate subtypes of the highly heterogeneous cancer using new technologies [8-10]. Multi-omic analyses and new systematic approach were utilized for the better classification and more sophisticated therapeutic strategy in the recent studies [11-16]. Nonetheless, only a few biomarkers have been identified to accurately predict prognosis and guide treatment decisions [17].
In this study, we aimed to develop a clinically applicable prognostic gene signature for patients with TNBC using whole transcription sequencing to classify the highly heterogeneous cancer more accurately. We combined weighted genes related to prognosis and validated the results using statistical analysis and meta-analysis. We also validated the discovered gene signature in a cohort of patients with TNBC at another institution.
Materials and Methods
1. Patients and specimens
The study included 184 patients with early-stage TNBC; 76 patients in the training cohort were from the National Cancer Center Korea (NCC, Goyang, Korea), and 108 patients in the validation cohort were from Samsung Medical Center (SMC, Seoul, Korea). All patients were eligible if they were ≥ 18-years-old with early-stage TNBCs (stage I-III), for whom a histological biopsy could be safely obtained and standard systemic chemotherapy (anthracycline and taxane) and locoregional treatment including surgery and radiation were applied. Tumor samples were identified as TNBCs according to the American Society of Clinical Oncology/College of American Pathologists (ASCO/CAP) guidelines [18,19]. The training cohort consisted of 15 patients who received neoadjuvant chemotherapy and 61 patients who underwent primary surgery followed by adjuvant chemotherapy for early-stage TNBC between March 2002 and August 2018. The validation cohort included 73 patients who had received neoadjuvant chemotherapy and 35 patients who received adjuvant chemotherapy after surgery between July 2011 and November 2017. In the validation cohort, 42 specimens of the neoadjuvant chemotherapy group were biopsy tissue before neoadjuvant chemotherapy, while the other specimens were surgical tissues. Tissues of training cohort were provided by NCC Bio Bank of National Cancer Center, Korea. All specimens were fresh-frozen.
2. Complete clinical information and outcome
Clinical data, including the date of diagnosis, clinical and surgical stages, response to neoadjuvant chemotherapy, recurrence, and survival, were collected from medical records. Invasive disease-free survival (iDFS) was defined as the time from diagnosis of primary breast cancer to invasive breast cancer recurrence or death from any cause and overall survival (OS) was defined as the time from diagnosis to death.
3. RNA sequencing and quality control
For the training cohort, sequencing libraries were prepared using fresh-frozen tissues with TruSeq Stranded mRNA LT Sample Prep Kit (Illumina Inc., San Diego, CA) following the manufacturer’s protocols. paired-end sequencing was conducted using the prepared cDNA library for RNA sequencing using an Illumina HiSeq 4000 sequencer (Illumina). In RNA sequencing quality control, artifacts including adaptor sequences, contaminant DNA, and PCR duplicates were eliminated to reduce the bias of sequencing data. After quality control of sequencing data, aligned reads were generated by mapping sequencing data on the reference genome using the HISAT2 program (GitHub, http://daehwankimlab.github.io/hisat2/). With generated aligned reads, transcript assembly was conducted using StringTie (https://ccb.jhu.edu/software/stringtie/). Based on the transcript quantification of each sample, expression levels were normalized to transcript length and depth of coverage. Through normalization, expression profiles were extracted as fragments per kilobase of transcript per million mapped reads (FPKM).
For the validation cohort, sequencing libraries were prepared using fresh-frozen tissues with TruSeq RNA Sample Prep Kit v2 (Illumina Inc.) following the manufacturer’s protocols. Sequencing of the RNA libraries was performed on a HiSeq 2500 sequencing platform (Illumina Inc.). After trimming poor-quality bases from the FASTQ files, we aligned the reads to the human reference genome (hg19) using STAR v.2.5 (GitHub, https://github.com/alexdobin/STAR) and estimated gene expression in terms of FPKM using RSEM v.1.3. The quality control of sequencing results was assessed using RNA-SeQC (v1.1.8).
For comparison between tumors and non-tumors, nontumor data were collected from Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/). GSE58135 was selected as the non-tumor group in GEO, and it had nontumor RNA sequencing data of 21 patients with TNBC [20]. In non-tumor data, data with a failed status or with the values of FPKM under 1.0×10–6 were excepted.
4. Differentially expressed gene and Cox regression analysis
By matching the genes of tumors and non-tumors, 10,856 genes were found in both groups. With 10,856 genes, the differentially expressed genes (DEGs) were analyzed. DEGs were screened to meet one of the following conditions: (1) statistically significant difference (Wilcoxon test, |fold change| > 1.5, adjusted p-value < 0.05) comparing tumors with nontumors and (2) statistically significant difference (Wilcoxon test, |fold change| > 1.5, adjust p-value < 0.05) comparing the patients who exhibited recurrence/metastasis after surgical resection with the patients without recurrence/metastasis. The previously screened DEGs were further shortlisted, matching with the gene significant in Cox regression analysis for recurrence/metastasis. Before combining the DEGs, we identified the Cox regression coefficient of each gene and weighted gene expression with the corresponding coefficient value [21].
5. Combination gene analysis
The 59 genes selected in the DEG analysis were subjected to combination analysis ranging from 2 to 10 genes. The combinations were formed by multiplying the regression coefficients of each correlating gene with its expression level and then summing them. Continuous Cox regression analysis was then performed to obtain p-values (p-values saturated in combinations of 8 to 10 genes). Subsequently, receiver operating characteristic (ROC) statistical analysis was conducted on the combined gene sets to calculate the area under the curve (AUC).
6. Pre-validation of the candidate gene signatures for recurrence by cross validation of machine learning
Candidate gene signatures (achieving p-value < 0.05, AUC > 0.90, sensitivity > 80%, and specificity > 80%) were ranked by k-fold cross validation to identify the optimal gene combination. The patients were randomly separated by 2 folds (training set and test set) 300 times [21].
7. Signal transduction pathway analysis based on meta-analysis
Signal transduction analysis was performed using the CBS Probe PINGS (Reg. No. 2008-01-129-000568, CbsBioscience, Daejeon, Korea) [21]. For gene signature validation, signal transduction was analyzed for pathways related to each patient’s DEGs compared recurrence/metastasis and non-recurrence/non-metastasis and for pathways related to gene signature. The genes were mapped to the signal transduction pathways obtained from the Kyoto Encyclopedia of Genes and Genomes (KEGG) database (http://www.genome.jp/kegg/). The top 10 signal transduction pathways were selected for each patient’s DEGs and for gene signature according to the weight of the number of interactions and interacting genes. Ten pathways related to each patient’s DEGs compared with GEO’s non-tumor and gene signature-related pathways were compared. For each signal transduction pathway selected in signal transduction pathway analysis, the gene interaction frequency ratio was computed, which is a score of interacting genes with signature genes in gene signature validation. By applying 100% gene interaction frequency to the highest probability of gene interaction within each signal transduction pathway, the top 10 high interaction frequency genes were selected. In addition, 10 high interaction frequency genes related to each patient’s DEGs and gene signatures related to high interaction frequency genes were compared.
8. Molecular subtype classification of TNBC
Molecular subtypes for both training and validation sets of data were primarily classified to six centroids using TNBC-type (http://cbc.mc.vanderbilt.edu/tnbc/) [22]. Using the results obtained from analysis, patients with TNBC were refined to four molecular subtypes such as basal-like 1 (BL1), basal-like 2 (BL2), mesenchymal (M), and luminal androgen receptor (LAR). Using Kaplan-Meier analysis, the prognostic power of TNBC-type-4 and the relationship between TNBC-type-4 and 10-gene signature were analyzed.
9. T-cell receptor diversity analysis
T-cell receptor (TCR) profiles were obtained using MiXCR 2.1.3 (GitHub, https://github.com/milaboratory/mixcr) using RNA sequencing data [23,24]. RNA sequencing data were aligned to all the IG/TCR loci. After two rounds of contig assembly, the V/J junctions of TCRs were extended. The assembled clonotypes were exported. TCR diversity was analyzed with the T cell receptor beta locus (TCRβ) using the Shannon index [25]. Using Kaplan-Meier survival (KM) analysis, the prognostic power of TCRβ diversity was analyzed.
10. Statistical analysis
Clinicopathological variables between the training and validation cohorts were evaluated using chi-square tests. Gene expression data were tested for normality using the Shapiro-Wilk test. As the data did not meet normality assumptions, significant differences between the responders and non-responders were evaluated using the Wilcoxon test. ROC curve analysis was used to determine the accuracy of threshold values for classifying recurrence/metastasis and non-recurrence/non-metastasis using gene signatures. KM curves were calculated using death and invasive disease as endpoints in iDFS and death in OS. The difference in KM curves was examined using the log-rank test, and the difference in hazard ratio was examined using Cox regression analysis. Candidate gene signatures were analyzed using Cox regression to understand the relationships between the recurrence/metastasis, classification, and clinicopathological variables. Significance was set at p < 0.05. All statistical analyses were performed using R v.3.4.3 software (R Development Core Team, https://www.r-project.org/).
Results
1. Clinical characteristics of patients
Of the 184 patients, 76 were in the training cohort and 108 in the validation cohort. The clinical characteristics of the patients in the training and validation cohorts are summarized in Table 1. Overall, the patients in the very young age group were not significantly different between the cohorts. Patients in the training cohort were more likely to have earlier-stage diseases than the validation cohort; however, the distribution of clinical stage among the patients was not different between the two groups. For adjuvant chemotherapy, the TAC (taxotere, adriamycin, and cyclophosphamide) regimen was used more in the validation cohort, probably because there were more advanced-stage patients. A schematic representation of the patients and samples is shown in S1 Fig.
2. DEG analyses of tumor vs. non-tumor, and relapsed vs. non-relapsed
In the DEG analysis of tumor versus non-tumor, 9,741 genes were significantly differentially expressed by more than 1.5-fold changes in 10,856 genes. DEG analysis of primary tumors between relapsed and non-relapsed patients revealed 136 out of 10,856 genes showing significant differences in expression by 1.5-fold. Subsequently, out of 10,856 genes, 584 genes were statistically significant in the single Cox analysis. In the DEG analysis in three ways, 59 DEGs overlapped (S2 Table).
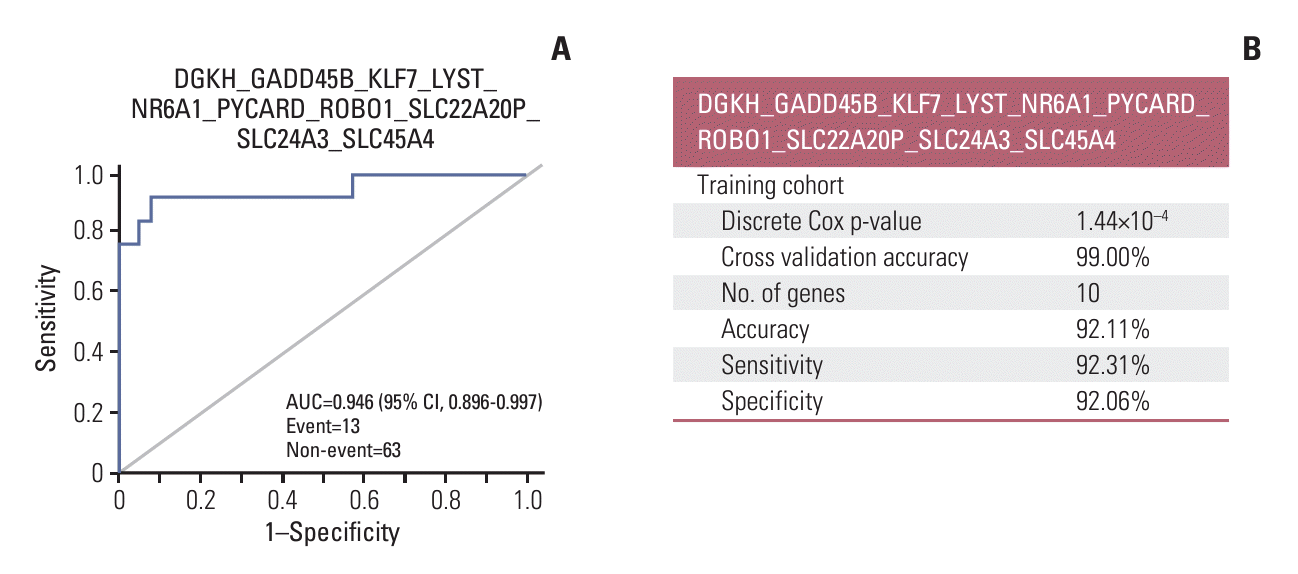
3. Candidate gene signatures by gene combination and selected gene signature by cross validation
The top 10 candidate gene signatures were ranked with the Continuous Cox p-value. Ten candidates showed values of 80 or higher in sensitivity, specificity, and accuracy. The prognostic gene signature was selected by meeting statistical criteria within subgroups of cohorts. Also the selected gene signature was DGKH_GADD45B_KLF7_LYST_NR6A1_PYCARD_ROBO1_SLC22A20P_SLC24A3_SLC45A4, showing 99.00% cross-validation accuracy, and it was statistically significant in the discrete Cox analysis. The risk score was calculated with a cut-off value of 5.959715 as follows: (0.818636×DGKH)+(0.018069×GADD45B)+(0.605352×KLF7)+(0.231666×LYST)+(1.305352×NR6A1)+(–0.052086×PYCARD)+(–0.196973×ROBO1)+(0.968759×SLC22A20P)+(0.098331×SLC24A3)+(0.311646×SLC45A4) (Table 2, Fig. 1).
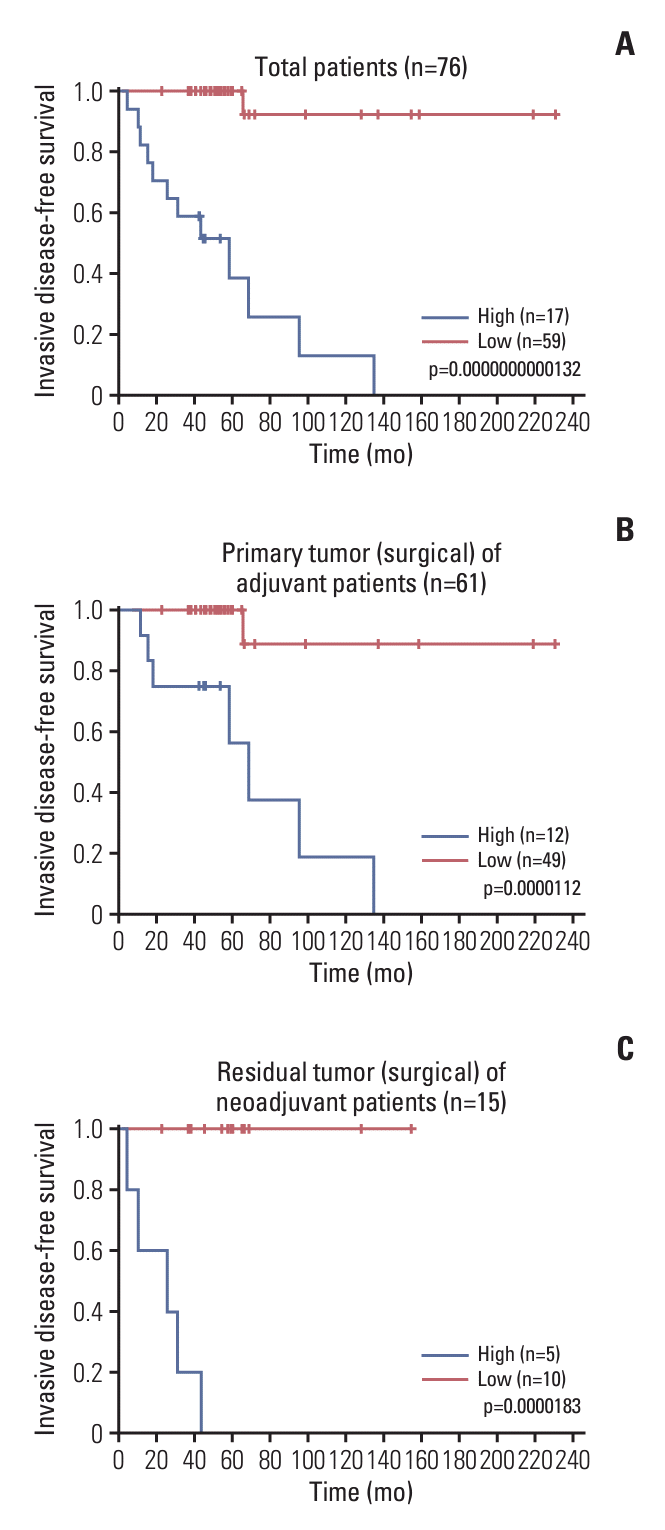
4. Prognostic significance of gene signature in the training cohort
During the median follow-up of 51.5 months (range, 4.6 to 230.8 months), patients with tumors with high-risk gene signatures (n=17) showed significantly shorter iDFS (median, 58.5 months; 95% confidence interval [CI], 25.8 to not reached) than those with low-risk signatures (n=59, median not reached, p=1.32e-11) in the overall population (Fig. 2A). Further analysis in the separate group of patients who underwent primary surgery and in the patients with residual tumors after neoadjuvant chemotherapy showed similar results. Among patients who received primary surgery and patients with residual tumors after neoadjuvant chemotherapy, the median iDFS in the high-risk group was 68.9 months (95% CI, 58.5 to not reached; p=1.12e-05) and 25.8 months (95% CI, 10.6 to not reached; p=1.83e-05), respectively, and the median iDFS in the low-risk group did not reach the median (Fig. 2B and C).
The impact of gene signature on OS of training set represented significant difference in total patients (p=0.00019). However, subgroup analysis represented significant impact only in neoadjuvant treated patients subgroup (S3 Fig.).
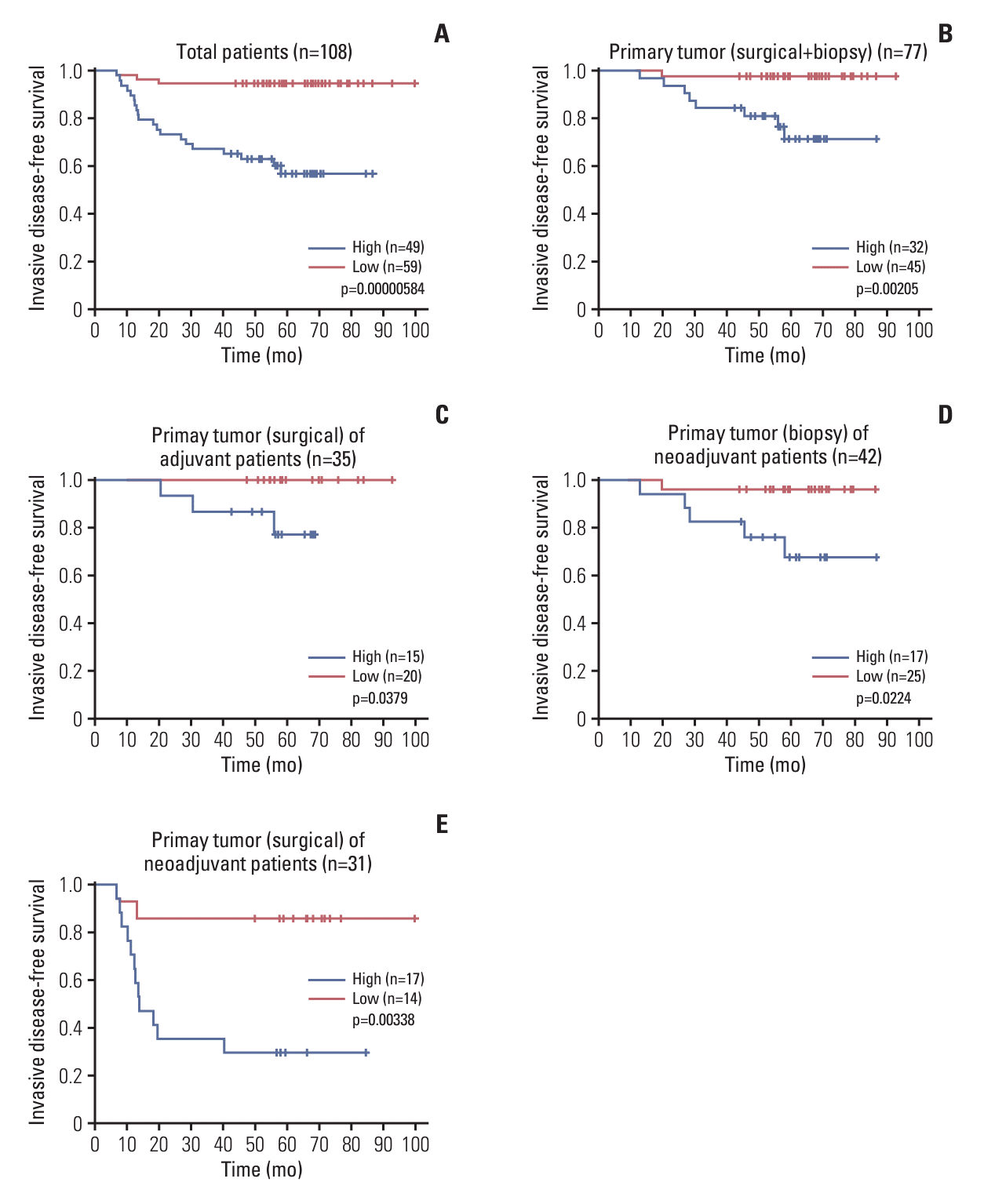
5. Prognostic significance of gene signatures in the validation cohort
Median follow-up time for validation cohort was 58.3 months (range, 6.6 to 99.8 months). In the overall validation cohort, although the median iDFS of the patients with high-risk genetic signatures was not reached, there was a significantly higher risk for recurrence or metastasis than patients with low-risk gene signatures (median iDFS not reached and p=5.84e-06 in log-rank test). When the patients were sub-divided according to the treatment sequence, the prognostic significance of the gene signatures in the surgical tissues from the patients who underwent primary surgery was consistent with the training cohort (p=0.0379); however, the median iDFS in the high-risk and low-risk groups was not reached yet. High-risk gene signatures were still valid in predicting the prognosis of patients with residual tumors after neoadjuvant chemotherapy; the median iDFS in the high-risk group was 13.6 months (95% CI, 12.2 to not reached), but it was not reached in the low-risk group (p=3.38e-03). Also, when the gene signatures were examined in the tissues obtained by core biopsy in the neoadjuvant chemotherapy group, prognostic significance in iDFS was statistically significant (p=0.0224) (Fig. 3).
In validation set, the gene signature impact on OS represented significance in total patients (p=0.00183). However, subgroup analysis represented significant impact in only primary tumor groups (p=0.0190) and marginal significance in neoadjuvant-treated patients group (p=0.0510) (S4 Fig.).
6. Relationships between 10-gene signature and other potential prognostic factors
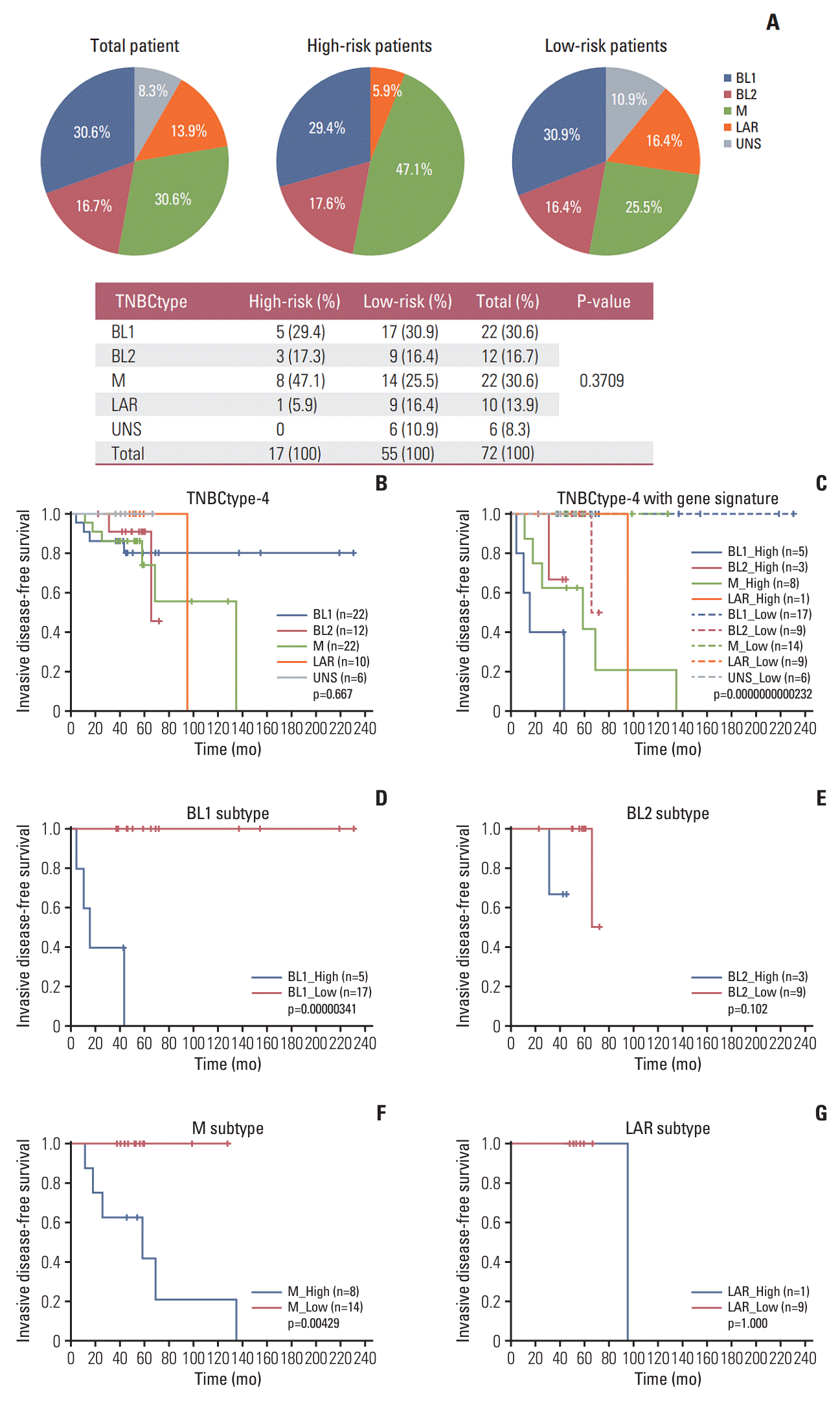
To investigate the interaction between the gene signature and other prognostic methods, specific prognostic values of the TNBC-type-4 were evaluated. In the TNBC-type-4 subgroup analysis, 76 patients with TNBC were classified into 22 patients of BL1 type (30.6%), 12 patients of BL2 type (16.7%), 22 patients of M type (30.6%), 10 patients of LAR type (13.9%) and six patients of unknown type (8.3%). There was no significant difference in terms of iDFS in the KM analysis by subtypes. However, patients could be further classified into high-risk and low-risk group by the gene signature in each subtype, respectively (Fig. 4). Among the TNBC subtypes, median iDFS of high-risk patients in 10-gene signature in BL1 (n=5, median iDFS was 15.5 months) and M (n=8, median iDFS was 58.5 months) subtypes were significantly inferior to those with low-risk patients (median not reached for both subtypes; p=3.41e-06 and p=4.29e-03 for BL1 and M, respectively). Additionally, we performed the TCRβ diversity analysis as a potential prognostic marker. With the cut-off that the highest point of the Youden index in ROC analysis (cut-off: 5.26), 35 patients belonged to the high diversity of TCRβ group and the rest of the patients had low diversity of TCRβ (n=41). However, the difference in iDFS according to the TCRβ diversity did not reach statistical significance (S5 Fig.).
7. Cox regression analysis of the selected gene signature
The prognostic impact of the selected gene signature (DGKH_GADD45B_KLF7_LYST_NR6A1_PYCARD_ROBO1_SLC22A20P_SLC24A3_SLC45A4) was investigated with Cox regression analysis. In the univariate Cox regression analysis, the gene signature was the most significant factor among the clinical factors and high score was strongly correlated with poor prognosis. TNM stage showed a marginal significance. In the multivariable Cox regression analysis with the gene signature, TNM stage, and TCRβ diversity, the gene signature only remained statistically significant (Table 3).
8. Signal transduction pathway analysis and high interaction frequency genes analysis for prognostic gene signature
Through the biological meta-analysis, we found gene signature and prognosis-related KEGG signal transduction pathways as well as high interaction frequency genes. Transduction pathway analysis revealed that the pathways in cancer, phosphoinositide 3-kinase–Akt signaling pathway, Alzheimer disease pathway, Human cytomegalovirus infection pathway, hepatitis C pathway, breast cancer pathway, and mitogen-activated protein kinase signaling pathway were related to the prognostic gene signatures and prognosis. In these pathways, KRAS, HRAS, and APP were the high interaction frequency genes related to gene signatures and prognosis (S6 Table).
Discussion
TNBCs are highly heterogeneous, making it challenging to predict prognosis and select appropriate treatments. Molecular categorization based on the comprehensive genetic signatures classified into distinct subtypes and therapeutic targets were suggested [14,26]. However, for the early-stage TNBCs, the introduction of immunotherapy was the only translation.
In this study, we aimed to identify and validate a 10-gene signature set based on the transcriptome of primary tumors for predicting the prognosis of patients with early-stage TNBC. Our analysis revealed that the gene signature set could better stratify patients with TNBC based on risk score than clinicopathologic parameters with an accuracy of 92.11% at a cut-off of 5.959715. We also validated the gene signature set was validated in patients with TNBC from another institution, demonstrating its objectivity.
Among the genes that make up the 10-gene set, some are related to immune responses (LYST and PYCARD), while others are related to the biology of cancer cells (GADD45B, KLF7, ROBO1, SLC45A4). The rest are novel genes with little evidence of a link to cancer (DGKH, NR6A1, SLC22A20P, SLC24A3). Detailed information on each gene that makes up the 10-gene set is presented in the S7 Table.
The objective of this study was to verify the 10-gene signatures in multivariate analysis by exploring additional biomarkers, in addition to traditional clinical variables. When our patient samples were analyzed according to the TNBC-type-4 subtyping [22], we observed a similar pattern of prognosis in our training cohort, based on subtypes (Fig. 4B). Further categorization of patients using the 10-gene signature revealed that patients with BL1 subtype and M subtype, but not BL2 and LAR subtypes, had a distinctly different prognosis (Fig. 4C-G). Using the gene score, we were able to differentiate patients with poor prognosis from those predicted to have a good prognosis for BL1 (5 out of 22, 22.7%). Among the total of 22 patients with the M subtype predicted to have poor risk, only eight (36.3%) were classified as high risk. However, the TCRβ diversity analysis showed that the mean level of TCRβ diversity was not significantly different between patients with relapsed and non-relapsed patients, and although patients with high TCRβ diversity (cut-off: 5.26) showed a trend for better iDFS, the difference was not statistically significant (S5 Fig.).
Interestingly, in the analysis using surgical specimens, the 10-gene signature discovered in this study significantly predicted the prognosis of patients with early-stage TNBC, regardless of whether the specimen was obtained from primary surgery or a residual tumor after neoadjuvant chemotherapy. In a recent analysis, 56% of cases showed a subtype change after neoadjuvant chemotherapy in patients with TNBC, and the most common change was from BL1 to M subtype [27]. Given that the BL1 and M subtypes account for the majority (55%-60%) of TNBC cases, the good performance of the 10-gene signature in these subtypes might have contributed to the consistent results in both pre-treatment and residual tumors.
Our approach to biomarker discovery differed from others in that we selected gene combinations that showed the best performance using combination analysis, which is a way to find optimal genes that can be used as biomarkers in large-scale analyses such as RNA sequencing. We then performed cross-validation as a pre-validation using a machine learning process. Through these procedures, we discovered a list of gene combinations with the least chance of failure in a separate validation cohort. Finally, a meta-analysis of the gene signature enabled us to demonstrate that the 10-gene signature has biological relevance in TNBC, not just as a list of genes with statistical power (S8 Fig.) [21].
We discovered a novel, 10-gene signature and validated it in a separate cohort of prospectively collected samples with regular follow-up data from a separate institution. There are several limitations in the application of the findings from this study. One of these limitations pertains to the heterogeneity of systemic chemotherapy regimens utilized, which were based on various clinical risk factors. It’s important to note that the patients involved in this study were enrolled prior to the implementation of findings from the CREATE-X or Keynote-522 studies into clinical practice [28,29]. Consequently, further investigation is warranted to assess the predictive potential of the gene signature concerning adjuvant capecitabine and/or immunotherapy. Given that the current standard treatment for early-stage TNBC involves a combination of pembrolizumab and chemotherapy as neoadjuvant therapy, additional research is necessary to ascertain the predictive role of the 10-gene signature in achieving pathologic complete remission or guidance for escalated treatment in high-risk patients.
In conclusion, our study identified a 10-gene signature as a potential prognostic biomarker for patients with early-stage TNBC. To be used as a biomarker in a risk-based approach to clinical practice, this gene signature should be further validated in prospective clinical studies involving new treatments such as capecitabine and immune checkpoint inhibitors are applied. Nonetheless, we suggest that our findings can contribute to solving diagnostic challenges in TNBC and to a step close to precision medicine for qualified patient care.
 XML Download
XML Download